
注:M为DL2000 Marker;1~6分别为YSp1、YSp2、BSp1、BSp2、YSp3、BSp3样品
Note:M, DL2000 Marker;1-6, YSp1,YSp2,BSp1, BSp2,YSp3,and BSp3 sample, respectively
图1 PCR扩增细菌16S rDNA琼脂糖凝胶电泳图
Fig.1 Agarose gelectrophoresis of bacterial 16S rDNA PCR production
摘要:以黄海和渤海春季刺参养殖池塘的沉积物、海水和刺参Apostichopusjaponicus肠道内容物中的细菌基因组DNA为模板,以细菌16S rDNA通用引物进行PCR扩增,构建16S rDNA文库并进行测序分析。结果表明:黄海和渤海养殖刺参肠道内容物、池塘沉积物和海水中共存在变形菌Proteobacteria、蓝细菌门Cyanobacteria、放线菌门Actinobacteria、拟杆菌门Bacteroidetes和厚壁菌门Firmicutes 5个门类的细菌类群,其优势类群均为变形菌(变形菌比例>41%),且在文库所含变形菌的4个亚门(Alphaproteobacteria、Betaproteobacteria、Gammaproteobacteria、Deltaproteobacteria)中,除黄海春季刺参养殖池塘海水文库(YSp3)中α-变形菌占优势外,其余5个文库均以γ-变形菌为优势亚门。
关键词:刺参;16S rDNA文库;变形菌
20世纪80年代以来,刺参Apostichopusjaponicus的增养殖在中国北方沿海蓬勃开展,然而随着养殖产业的快速发展,刺参病害问题日益突出,成为制约该养殖产业持续发展的瓶颈[1]。微生物在养殖生态系统的物质循环和能量流动中发挥重要作用,养殖生物与其有着密切的关系,常常是引起养殖对象致病的病原或条件致病性病原。研究表明,细菌性疾病在刺参养殖中非常普遍[2],因此,对养殖环境中微生物多样性的研究具有重要意义。
自然界中绝大部分细菌不可培养,传统纯培养法只能培养出较少细菌种类,不能真实反映菌群结构[3]。现代分子生物学方法为微生物多样性的研究提供了有效手段,16S rDNA法不依赖于培养出的菌株,而是以样品中细菌的DNA为研究目标进行克隆分析,从而能更真实地反映所检测样品的菌群结构及其多样性。国内外已有学者利用16S rDNA克隆文库法对细菌结构进行分析,李革雷等[4]研究了3种养殖模式水体中的细菌多样性,Sekigucui等[5]研究了鄱阳湖湖口水体的菌群结构,Julian等[6]分析了深海沉积物的菌群多样性,郑艳玲等[7]研究了崇明东滩夏冬两季沉积物中的菌群结构,丁君等[8]检测了刺参消化道的细菌种类。本研究中,以中国北方黄海和渤海刺参养殖区的海水、沉积物和刺参肠道内容物中细菌基因组为模板,构建 16S rDNA克隆文库,分析北方养殖刺参内外生境菌群的多样性以及黄、渤海刺参养殖池塘和刺参肠道中的菌群结构,旨在为构建中国北方刺参养殖池塘菌库提供重要依据。
1.1材料
试验用刺参样品于2012年5月分别采集于黄海、渤海养殖区某刺参养殖场。
1.2方法
1.2.1 样品的采集 样品的采集参照王轶南等[9]的方法。黄海刺参肠道内容物、池塘沉积物、海水样品分别标记为YSp1、YSp2、YSp3;渤海刺参肠道内容物、池塘沉积物、海水样品分别标记为BSp1、BSp2、BSp3。
1.2.2 样品DNA的提取 刺参肠道内容物、池塘沉积物和海水样品中细菌DNA的提取参照王轶南等[9]的方法。
1.2.3 PCR扩增16S rDNA 采用16S rDNA通用PCR引物27F(5′ AGAGTTTGATCCTGGCTCAG3′)和1492R(5′ GGTTACCTTGTTACGACTT 3′)对样品DNA进行扩增。PCR反应体系(共25 μL):2×Power Taq PCR MasterMix 12.5 μL,上、下游引物各1 μL,模板1 μL,用双蒸水补至25 μL。PCR反应程序:95 ℃下预变性2 min;94 ℃下变性30 s,52 ℃下退火30 s,72 ℃下延伸90 s,共进行30个循环;最后在72 ℃下再延伸5 min,4 ℃下保存备用。PCR产物经10 g/L琼脂糖凝胶电泳确定目的条带后,切胶回收纯化。
1.2.4 16S rDNA克隆文库的构建 经纯化后的PCR产物连接到载体pMD19-T中,16 ℃下连接30 min;将连接产物转化至感受态细胞中,在不含氨苄青霉素(Amp)的液体LB培养基中培养1 h后,涂布于含有X-gal、IPTG和Amp的LB培养基上;随机挑选100个左右白色克隆子送至上海英俊生物技术有限公司进行测序。
1.2.5 文库分析 用VecScreen程序去除载体序列,用DECIPHER在线软件[10]进行嵌合体检验。用Mothu软件[11]对所得有效序列进行分析,以97%相似性为标准划分操作分类单元(operational taxonomic unit,OTU)。用RDP-Classifier在线软件[12](http://rdp.cme.msu.edu/classifier/classifier. jsp)对所得有效序列进行菌属鉴定。基于OTU,用SPADE软件[13]计算YSp1、YSp2、YSp3、BSp1、BSp2、BSp3克隆文库的覆盖率、香农指数、辛普森指数。将每个OTU中的代表序列利用GenBank数据库中Blast程序进行相似性比对,并挑选相近序列。采用Neighbor-joining法[14]并将自展值设为1000次[15],应用Mega 5.22软件构建系统发育树。
2.1 16SrDNA扩增检测
样品DNA PCR扩增后经琼脂糖凝胶电泳检测(图1),扩增条带单一,扩增片段大小符合预期结果(约1500 bp),表明扩增效果较好。
注:M为DL2000 Marker;1~6分别为YSp1、YSp2、BSp1、BSp2、YSp3、BSp3样品
Note:M, DL2000 Marker;1-6, YSp1,YSp2,BSp1, BSp2,YSp3,and BSp3 sample, respectively
图1 PCR扩增细菌16S rDNA琼脂糖凝胶电泳图
Fig.1 Agarose gelectrophoresis of bacterial 16S rDNA PCR production
2.2多样性分析
6个克隆文库中的克隆序列经载体去除和嵌合体检测后,共得到540条有效克隆序列,所有序列可归为37个OTUs。6个文库的覆盖率为86.5%~99.0%。其中黄海春季刺参养殖池塘海水(YSp3)文库的香农指数最大(2.986),辛普森指数最小(0.099 10),群落多样性较高(表1)。
表1黄、渤海春季刺参肠道及养殖池塘的细菌多样性
Tab.1FloradiversityofbacterialcommunitiesfromtheintestineandculturepondsofseacucumberApostichopusjaponicusfromYellowSeaandBohaiinspring
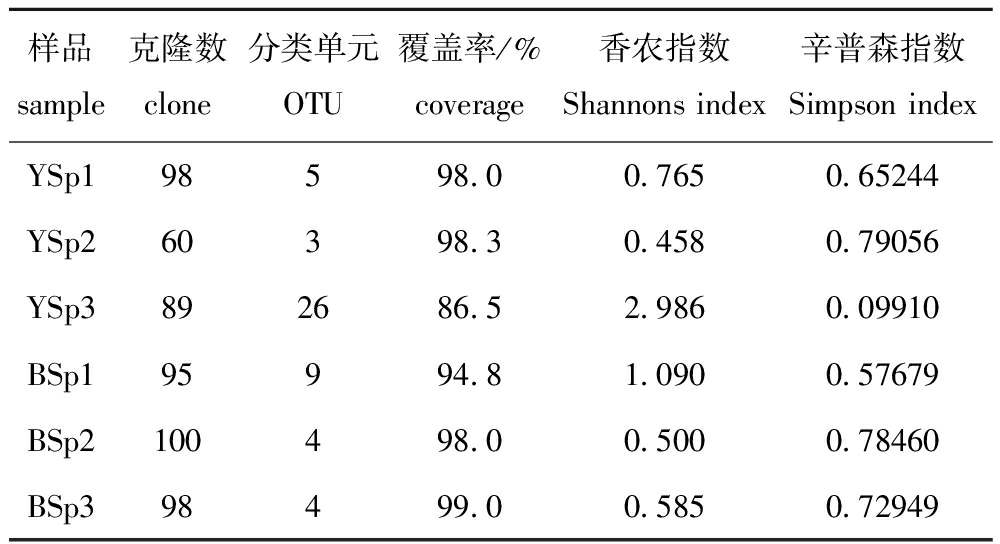
样品sample克隆数clone分类单元OTU覆盖率/%coverage香农指数Shannonsindex辛普森指数SimpsonindexYSp198598 00 7650 65244YSp260398 30 4580 79056YSp3892686 52 9860 09910BSp195994 81 0900 57679BSp2100498 00 5000 78460BSp398499 00 5850 72949
2.3系统进化树分析
黄、渤海春季养殖刺参内外生境中,6个文库序列可归为5个门类(图2):变形菌Proteobacteria、蓝细菌门Cyanobacteria、放线菌门Actinobacteria、拟杆菌门Bacteroidetes和厚壁菌门Firmicutes,其中变形菌均为6个文库的优势门类(变形菌比28例>41%);在文库所含变形菌的4个亚门(Alphaproteobacteria、Betaproteobacteria、Gammaproteobacteria、Deltaproteobacteria)中,除YSp3文库中α-变形菌占优势外, 其余文库中均以γ-变形菌占优势。基于RDP-Classifier在线软件分析,γ-变形菌中能够分到属水平的共有3类:Stenotrophomonas、Acinetobacter、Escherichia/Shigella。其中Stenotrophomonas在6个文库中均有分布;除YPs3文库外,Acinetobacter在其余5个文库中均有分布;Escherichia/Shigella主要分布于YSp1、YSp2、BSp1、BSp3文库中。此外,渤海春季养殖刺参肠道内容物和池塘沉积物归为厚壁菌门的克隆序列,经进一步分类分析均属于Bacillus属。
黄海春季文库中,YSp1和YSp2文库中菌群类型组成相似,两者与YSp3文库中菌群类型组成差异较大;渤海春季文库中,BSp1、BSp2和BSp3文库中菌群类型组成相似(图3)。
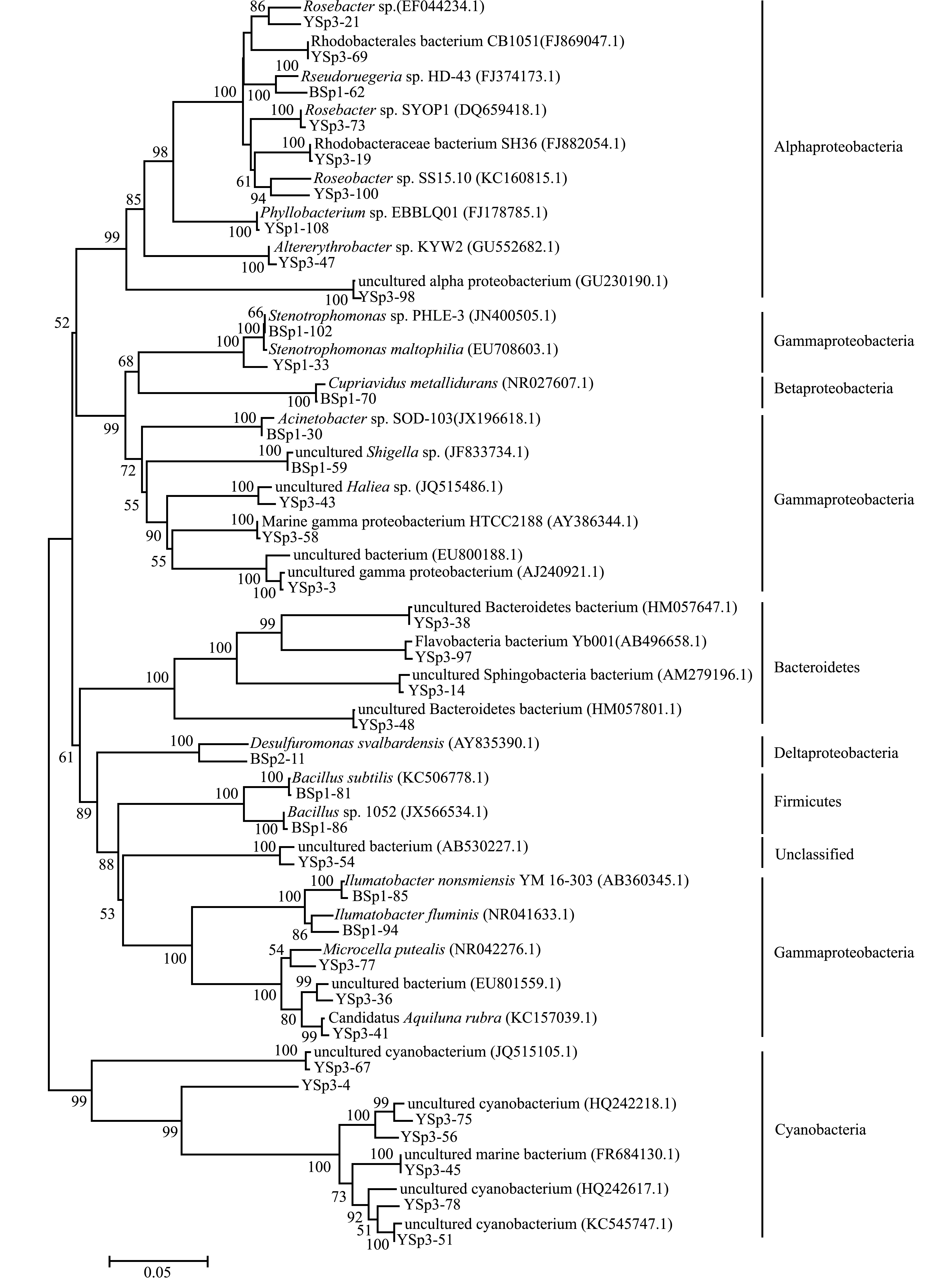
图2 黄、渤海春季刺参肠道及养殖池塘细菌16S rDNA序列系统发育树
Fig.2 Phylogenetic tree of bacterial 16S rDNA sequences from the intestine and culture ponds of sea cucumber Apostichopus japonicus from Yellow Sea and Bohai in spring
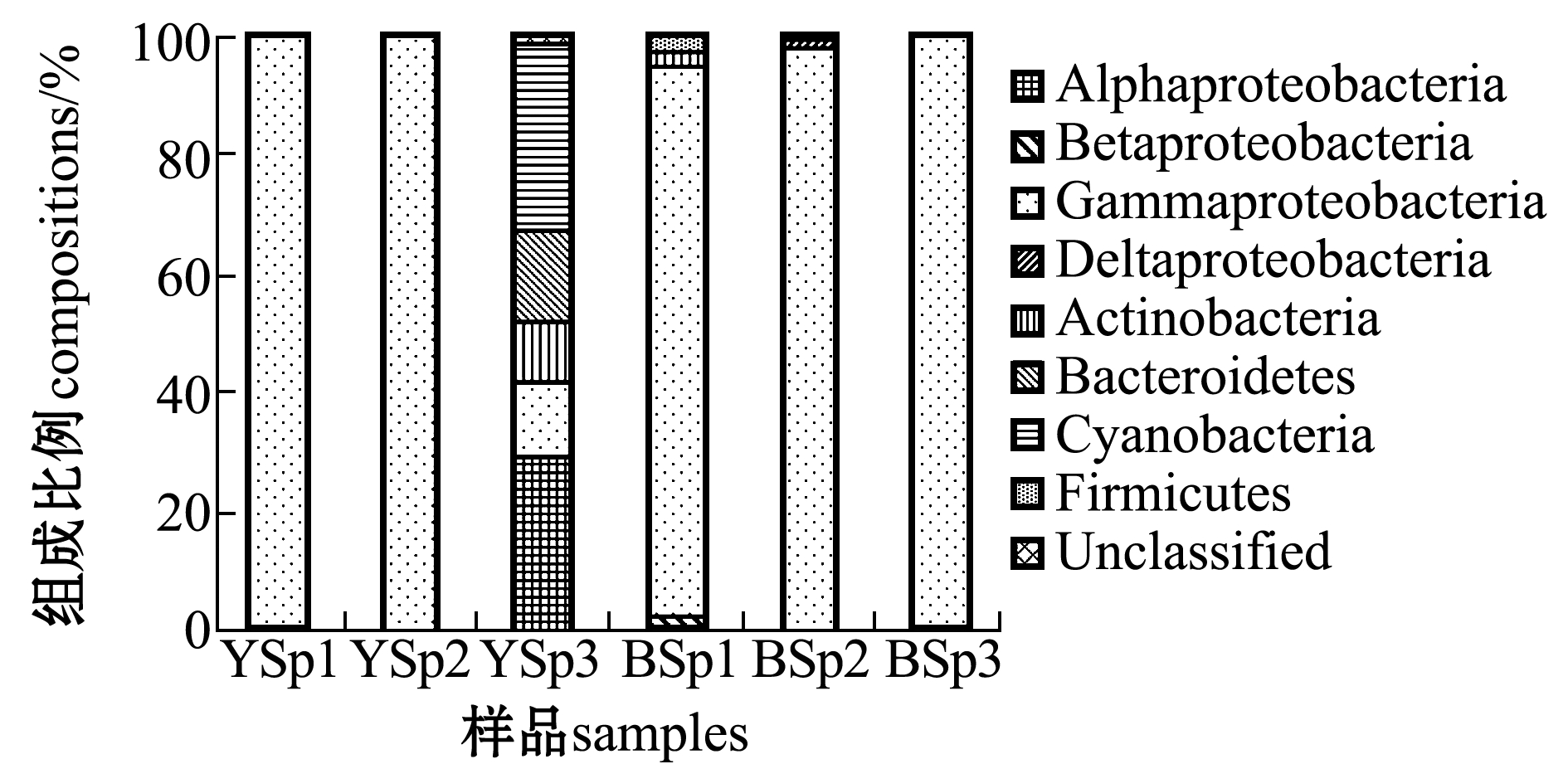
图3 黄、渤海春季刺参肠道及养殖池塘的细菌组成
Fig.3 Community compositions of bacteria in the intestine and culture ponds of sea cucumber Apostichopus japonicus from Yellow Sea and Bohai in spring
3.1海水细菌文库菌群的结构分析
Jack等[16]对普利茅斯沿海地区的海水进行调查,发现在海水文库中的优势菌为变形菌;关晓燕等[17]利用16S rDNA PCR-DGGE技术对辽宁省大连、普兰店湾刺参养殖环境中菌群多样性的研究中指出,变形菌是养殖水体中的优势菌。与前人研究结果相一致,本研究中黄海和渤海春季刺参养殖池塘海水文库均以变形菌为优势类群。比较而言,两地的养殖池塘海水中菌群类型差异较大,黄海春季刺参养殖池塘海水文库中除变形菌门外,还含有蓝细菌门、放线菌门、拟杆菌门;渤海春季刺参养殖池塘海水文库中仅能检测到变形菌门类细菌,且在变形菌类群中以γ-变形菌为优势类群。养殖环境不同,可能是导致其菌群结构不同的主要原因。
3.2养殖池塘沉积物细菌文库菌群的结构分析
本研究结果表明,黄海和渤海刺参养殖池塘沉积物文库中的优势类群均为变形菌门,这一结果与近年来有关海洋沉积物中微生物多样性的报道相一致。葛良法[18]的研究显示,海水养殖池塘沉积物中存在丰富的细菌类群,变形杆菌为优势类群;王丽萍等[19]的研究表明,天津新港区潮间带地区的沉积物中变形杆菌为优势菌;白洁等[20]的研究表明,黄海西北部各站位沉积物中变形菌门类为优势菌。γ-变形菌具有很强的适应性,它们在不同的生态系统中有着广泛的分布。闫法军[21]在研究刺参养殖池塘生态系统微生物结构时发现,刺参池塘底泥中γ-变形菌占绝对优势。这一结果在本研究中也得到证实。Urakawa等[22]的研究表明,海水中占较大比例的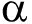 -变形菌很少在海洋沉积物中被发现。本研究结果与其相一致,刺参养殖池塘沉积物中均未发现-变形菌。
-变形菌很少在海洋沉积物中被发现。本研究结果与其相一致,刺参养殖池塘沉积物中均未发现-变形菌。
芽孢杆菌是首先应用于养殖生产中的益生菌[23],益生菌对养殖刺参的影响目前已有诸多研究。本研究中仅在渤海刺参养殖池塘沉积物文库中发现一条克隆序列属于芽孢杆菌,在黄海刺参养殖池塘沉积物文库中未检测到。而王亚南等[24]研究发现,福建某近海海水养殖场底泥细菌的优势菌为芽孢杆菌;李彬等[25]研究也发现,冬季青岛刺参养殖池塘沉积物中的优势菌为芽孢杆菌。推测这可能是养殖环境和采样地点不同造成的。
3.3刺参肠道内容物文库菌群结构及其与外生境菌群结构的比较分析
高菲等[26]利用PCR-DGGE技术分析刺参肠道内容物细菌的群落组成时发现,刺参前肠、中肠和后肠内容物的优势菌群均为γ-变形菌。本研究中也发现,黄海和渤海春季养殖刺参肠道内容物文库中γ-变形菌为优势类群,分别占两文库的98.02%和92.63%。高菲等[26]所获得的13条序列中,12条与之亲缘关系最近的序列来自从海洋环境中获得的细菌克隆,表明刺参消化道的细菌群落可能直接或间接来源于刺参的栖息环境。本研究中所建文库中,变形菌均为6个文库的优势门类。相较而言,黄海和渤海养殖刺参的肠道内容物与相应地点的刺参养殖池塘海水文库菌群结构差异较大,在黄海刺参养殖池塘海水文库中除变形菌外,还检测到放线菌门、拟杆菌门、蓝细菌门细菌,且所占比例较大,但这3种菌在黄海刺参肠道内容物文库中均未检测到;在渤海刺参肠道内容物文库中除变形菌外,还检测到放线菌门和厚壁菌门细菌,但在渤海刺参养殖池塘海水文库中未检测到。与此不同的是,本研究中发现,黄海和渤海刺参肠道内容物文库和刺参养殖池塘沉积物文库中变形菌均为优势类群,结构也比较相似。推测原因,可能是由于刺参摄食无选择性,在池塘底层摄入大量的底泥、有机物质和细菌,加之刺参肠道中有稳定的细菌菌群辅助消化[27],刺参摄入的一些细菌不能被消化,而是在肠道中定植下来。
参考文献:
[1] Deng H,He C B,Zhou Z C,et al.Isolation and pathogenicity of pathogens from skin ulceration disease and viscera ejection syndrome of the sea cucumberApostichopusjaponicus[J].Aquaculture,2009,287:18-27.
[2] 隋锡林,邓欢.刺参池塘养殖的病害及防治对策[J].水产科学,2004,23(6):22-23.
[3] Widada J,Nojiri H,Omori T.Recent developments in molecular techniques for identification and monitoring of xenobiotic-degrading bacteria and their catabolic genes in bioremediation[J].Appl Microbiol Biotechnol,2002(6):45-59.
[4] 李革雷,陈昌福,高宇,等.3种养殖模式水体中细菌多样性研究[J].华中农业大学学报,2012,31(3):381-390.
[5] Sekigucui H,Watanabe M,Nakahara T,et al.Succession of bacterial community structure along the Changjiang River determined by denaturing gradient gel electrophoresis and clone library analysis[J].Appl Environ Microbiol,2002,68(10):5142-5150.
[6] Julian R,Andrew J,Barry A,et al.Methanogen and bacterial diversity and distribution in deep gas hydrate sediments from the Cascadia Margin as revealed by 16S rRNA molecular analysis[J].Microbiology Ecology,2001,34(3):221-228.
[7] 郑艳玲,侯立军,陆敏,等.崇明东滩夏冬季表层沉积物细菌多样性研究[J].中国环境科学,2012,32(2):300-310.
[8] 丁君,李娇,王姮,等.利用16S rDNA方法检测刺参消化道细菌种类[J].海洋环境科学,2010,29(2):250-254.
[9] 王轶南,朱世伟,常亚青.刺参肠道及养殖池塘菌群组成的PCR-DGGE指纹图谱分析[J].渔业科学进展,2010,31(3):119-122.
[10] Wright E S,Yilmaz S,Noguera D R.DECIPHER,a search-based approach to chimera identification for 16S rRNA sequences[J].Appl Environ Microbiol,2012,78(3):717-725.
[11] Svhloss P D,Westcott S L,Ryabin T,et al.Introducing Mothur:open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Appl Environ Microbiol,2009,75(23):7537-7541.
[12] Wang Q,Garrity G M,Tiedje J M,et al.Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Appl Environ Microbiol,2007,73(16):5261-5267.
[13] National Tsing Hua University,TAIWAN.Program SPADE (Species Prediction And Diversity Estimation)[EB/OL].[2013-01-01].http://chao.stat.nthu.edu.tw.2010.
[14] Saitou N,Nei M.The Neighbor-joining method:a new method for reconstructing phylogenetic trees[J].Molecular Biology and Evolution,1987,4(4):406-425.
[15] Felsenstein J.Confidence limits on phylogenies:an approach using the bootstrap[J].Evolution,1985,39(4):783-791.
[16] Jack A G,Joshua A S,Gregory C.Defining seasonal marine microbial community dynamics[J].The ISME Journal,2012,6(2):298-308.
[17] 关晓燕,周遵春,姜冰,等.DGGE分析不同盐度仿刺参养殖环境中菌群多样性[J].水产科学,2011,30(5):276-280.
[18] 葛良法.海水及海水养殖场沉积物中微生物遗传多样性研究[D].广州:中山大学,2004.
[19] 王丽萍,郑丙辉.天津新港区潮间带沉积物细菌群落结构[J].应用与环境生物学报,2011,17(3):303-306.
[20] 白洁,李海艳,张健,等.黄海西北部沉积物中细菌群落16S rDNA多样性解析[J].中国环境科学,2009,29(12):1277-1284.
[21] 闫法军.刺参养殖池塘生态系统微生物结构域功能研究[D].青岛:中国海洋大学,2013.
[22] Urakawa H,Kita-Tsulcamota K,Ohwada K.Microbial diversity in marine sediments from Sagami Bay and Tokyo Bay,Japan,as determined by 16S rRNA gene analysis[J].Microbiology,1999,145(11):3305-3315.
[23] 张艳,李秋芬,王印庚,等.益生菌的研究现状及其在海水养殖中的应用[J].海洋水产研究,2005,26(6):83-87.
[24] 王亚南,彭志英,刘双江.养殖场底泥中芽孢杆菌属细菌的生态学研究[J].湛江海洋大学学报,2004,24(4):23-27.
[25] 李彬,荣小军,廖梅杰,等.冬季刺参养殖环境与肠道内细菌菌群研究[J].海洋科学,2010,34(4):64-69.
[26] 高菲,孙慧玲,许强,等.刺参消化道内含物细菌群落组成的PCR-DGGE分析[J].中国水产科学,2010,17(4):671-680.
[27] Clifford C,Walsh J,Reidy N,et al.Digestive enzymes and subcellular location of disaccharidases in some echinoderms[J].Comp Biochem Physiol,1982,71B:105-110.
Abstract:The 16S rDNA genes of bacteria were amplified and 16S rDNA clone libraries were constructed based on the total DNA extracted from the intestine of and water and sediments in culture ponds of sea cucumberApostichopusjaponicusfrom Yellow Sea and Bohai in spring. The results showed that the bacteria in the intestine and culture pond were composed of Proteobacteria, Cyanobacteria, Actinobacteria, Bacteroidetes and Firmicutes, with dominant Proteobacteria (>41%)in the intestine and culture ponds. Among the four subphyla of Proteobacteria(Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria and Deltaproteobacteria), Alphaproteobacteria was dominant in the library of seawater in sea cucmber culture ponds from Yellow Sea in spring, while Gammaproteobacteria was dominant group in others.
Key words:Apostichopusjaponicus; 16S rDNA clone library; Proteobacteria
DOI:10.3969/J.ISSN.2095-1388.2014.06.006
文章编号:2095-1388(2014)06-0572-05
收稿日期:2014-03-28
基金项目:国家海洋公益项目(201105007-2);辽宁省教育厅优秀人才支持计划项目(LJQ2011073)
中图分类号:S917.1
文献标志码::A