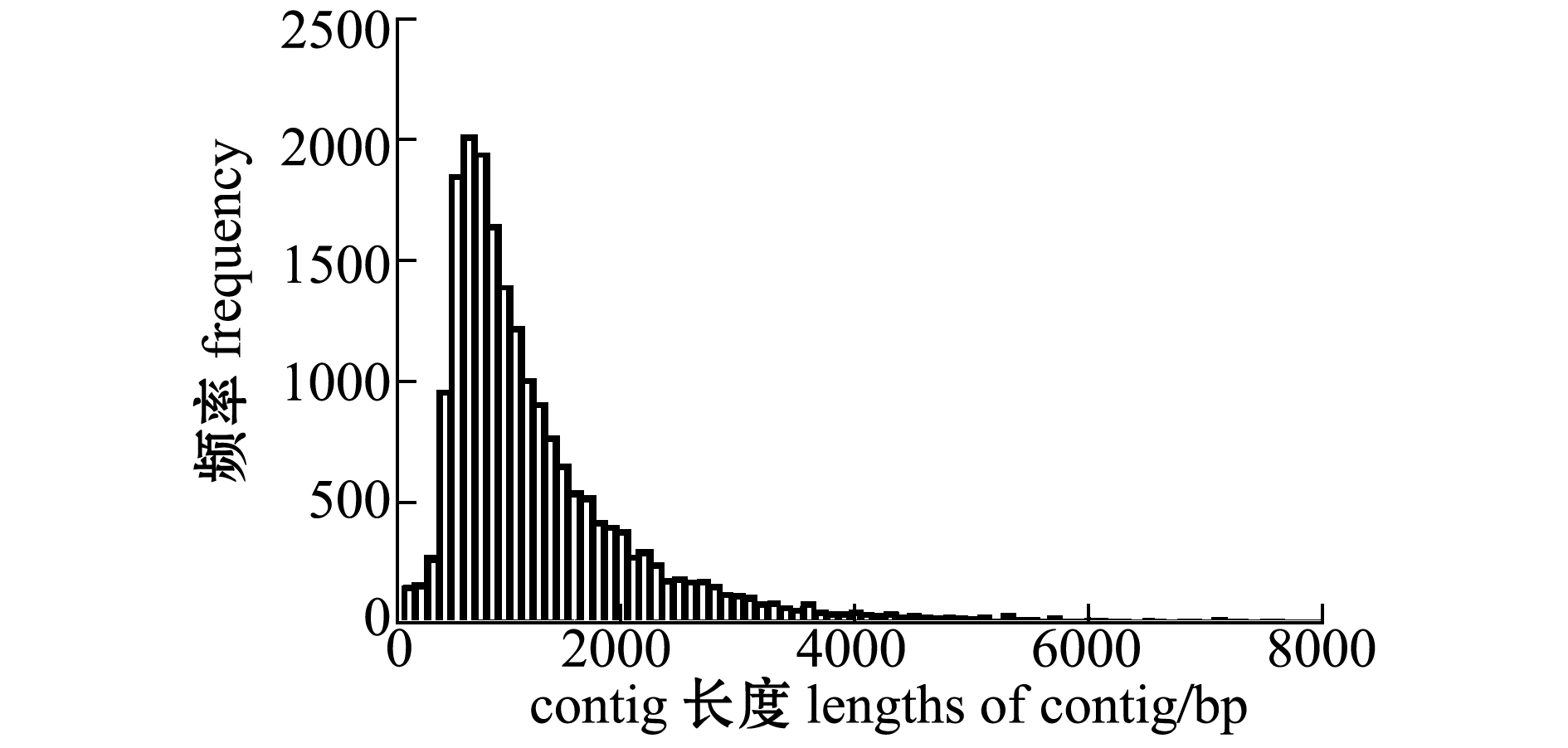
图1 鲮转录组contig长度的分布图
Fig.1 Length freguency of assembled transcriptome contigs in mud carp
摘要:为满足标记辅助育种的要求,通过454测序平台首次开展了鲮Cirrhinamolitorella全鱼转录组深度测序,并用Newbler等软件进行数据精细分析。结果表明:共获得了1 297 479条reads,总碱基数为486 586 191 bp,组装后得到19 962条contigs,平均长度为1269 bp,N50为1509 bp。基因功能注释研究共获取了10 577 个特异蛋白,根据特异蛋白注释结果进行GO分析,有7314条contigs有GO注释,包含5381个特异蛋白;采用GO 功能分类工具可将已注释转录物序列划分为分子功能、生物途径和细胞成分3类,为下一步开展生长等性状相关基因功能验证研究提供丰富的序列资源;共鉴定出5931个具有完整的ORF的全长cDNA序列,并且鉴定出2438个微卫星和5014个SNP位点。本研究中,还建立了鲮转录组数据库和网站,方便同行随时调取数据,这为深入开展鲮分子标记辅助的遗传育种、种群遗传学和资源评估等研究提供了丰富的标记资源。
关键词:鲮;转录组;高通量RNA测序;从头组装
鲮Cirrhinamolitorella俗称土鲮、鲮公、花鲮,是中国珠江流域地区的特有种,华南地区“四大家鱼”(鲢Hypophthalmichthysmolitrix、鳙H.snobilis、草Ctenopharyngodonidella、鲮)之一,仅在广东省年养殖产量就在20万t左右。鲮肉质细嫩、味鲜美、产量大、价格适中,是市场的畅销水产品。鲮也可入药,具有健筋骨、活血行气、逐水利温之功效。目前,生长速度慢和不耐寒等问题是鲮产业发展的瓶颈,快速生长的鲮品系不仅会提高传统养殖地区渔民的养殖积极性,也可以推广到生长期较短的北方地区,能极大地提高鲮在中国淡水渔业中的地位。所以,选育快速生长的鲮新品系是解决鲮产业发展中关键问题的有效途径。
近年来,珠江水产研究所通过对野生鲮资源及其生长参数进行调查,在西江群体中筛选出一个早期生长较快的群体,有效地进行了鲮的保种和扩繁,可作为进一步选育的基础群。然而,传统家系选育方法历时长、工作量大,而分子标记辅助育种则可以大大节省时间和劳动力,是一种快速有效的育种措施,但受限于鲮基因组信息较少,迄今为止,仅有少量微卫星和零星单核苷酸多态性(SNP)标记在种群遗传分析中进行了初步应用[1-3],远远不能满足标记辅助育种的要求,亟须找出一种快速、大量地获得鲮遗传信息的途径。此外,对鲮营养、生理生化等方面的研究,也亟须获取鲮相关的功能基因,而目前仅仅依赖从近缘模式鱼类斑马鱼Daniorerio基因组获取相关序列信息,然后进行繁琐的分子克隆实验才能获取相关基因和序列。因此,尽快建立鲮转录组数据库具有重要的意义。鉴于此,本研究中开展了鲮的首个高通量转录组研究,采集代表性鲮种群中多个个体的组织样本,采用第二代基因组测序技术进行深度的转录组测序,利用生物信息学分析流程,系统地进行了序列清洗、基因拼接、全长序列获取、基因功能注释、重复序列和元件分析评估、微卫星和SNP位点挖掘等分析研究,并建立了鲮转录组数据库和网站,旨在方便同行随时调取数据,实现数据共享。
1.1材料
试验用鲮采自珠江水产研究所实验基地,取生长良好的鲮幼鱼10尾,体长约为3 cm,体质量约为3 g。
1.2方法
1.2.1 样品的采集和总RNA的抽提 将10尾鱼迅速置于液氮预冷的研钵中,边加液氮边研磨至粉末状,混合后使用Invitrogen公司的TriZol试剂进行总 RNA抽提。使用安捷伦生物分析仪2100和紫外分光光度仪检测总RNA 的质量和数量。
1.2.2 cDNA文库的构建和测序 取100 ng RNA样品(10条鱼混合样),利用罗氏公司的Ovation RNA-Seq试剂盒 (NuGEN Technologies,SanCarlos,CA)合成cDNA,通过末端修复、连接接头和纯化,获得鲮样品的cDNA文库。将该cDNA文库用454平台测序,运行通量为1个run。
1.2.3 de novo拼接及数据分析 采用454测序技术,原始的测序数据被保存为454特有的SFF文件格式,由于没有可用的参考基因组数据,使用Newbler 2.8的-cdna模式进行de novo拼接,并使用-vt参数去除载体序列,用-vs参数去除核糖体RNA序列,最终获取初步拼接结果,并保留拼接后大于100 bp的contig。
1.2.4 SNP分析 为了进行SNP鉴定和分析,将Newbler清洗后的SFF格式文件通过PERL脚本转换成fastq文件,以Newbler软件拼接的转录组结果作为参考序列,采用BWA和SAMtools软件对转录组SNP进行鉴定,测序深度大于10且测序质量值大于20作为过滤阈值,其余参数为默认值。
1.2.5 重复序列分析及微卫星鉴定 对侧翼长度大于50 bp的微卫星序列采用Msatfinder 2.0.9程序进行微卫星鉴定。其中鉴定二、三、四、五、六核苷酸的重复,阈值分别被设为8、5、5、5、5。
1.2.6 功能注释 使用在线生物信息学分析软件BlastX将组装得到的转录组数据与NCBI的非冗余蛋白数据库、斑马鱼蛋白数据库和UniProt蛋白数据库进行比对,获得同源蛋白匹配结果,e-value 阈值设定为1E-5。为进一步通过NCBI Entrez Gene数据库和Ensembl斑马鱼基因组数据的注释,通过同源比对进行GO注释,并使用WEGO进行生物过程、分子功能和细胞成分的富集分析。
2.1转录组组装
采用454测序技术获得了鲮转录组的数据。对原始数据进行统计,共获得1 297 479条reads,总碱基数为486 586 191 bp。最大的reads长度为1195 bp,最小的reads长度为40 bp,平均reads长度为375 bp。
鲮的转录组拼接结果显示,共得到19 962条contigs,平均contig长度为1269 bp,N50为1509 bp,最大的contig长度为20 289 bp,最小的contig长度为101 bp。对每条contig长度统计相应的contig数量,结果如图1所示,contig长度主要分布在600~800 bp。
图1 鲮转录组contig长度的分布图
Fig.1 Length freguency of assembled transcriptome contigs in mud carp
2.2功能注释及GO分析
将组装的contig与NCBI的非冗余蛋白库(NR)、UniProt蛋白库和Ensembl斑马鱼蛋白库进行BlastX比对。结果显示,总共有13 657条contigs在NR蛋白数据库中被比对出,其中包括10 577个特异蛋白(表1)。
表1NR、Uniprot和Zebrafish数据库的BlastX比对结果
Tab.1BlastXsearchresultsagainstNR,UniprotandZebrafishdatabase
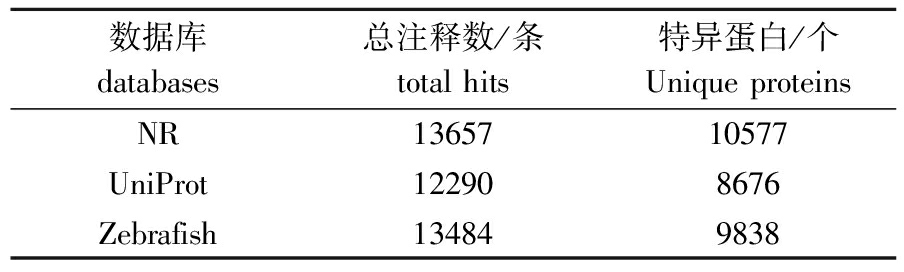
数据库databases总注释数/条totalhits特异蛋白/个UniqueproteinsNR1365710577UniProt122908676Zebrafish134849838
根据特异蛋白注释结果进行GO分析,结果有7314条contigs有GO注释,包含5381个特异蛋白。将注释信息整理成WEGO所需的输入文件,共分为分子功能(molecular function)、生物途径(biological process)和细胞成分(cellular component)3个大类(图2)。在生物过程中,与细胞过程(cellular process)(GO:0009987,GO注释条目2694,GO注释条目占总条目的比例为50.1%)和代谢过程(metabolic processes)(GO:0008152,GO注释条目2133,39.6%)相关的基因产生了显著的富集。对于分子功能,连接(binding)(GO:0005488,GO注释条目2716,50.5%)是最为主要的成分,其次是催化活性(catalytic activity)(GO:0003824,GO注释条目1793,33.3%);而细胞(cell)(GO:0005623,GO注释条目2675,49.7%)和细胞组分(cell part)(GO:0043226,GO注释条目2675,49.7%)是最具代表性的类别的细胞成分。
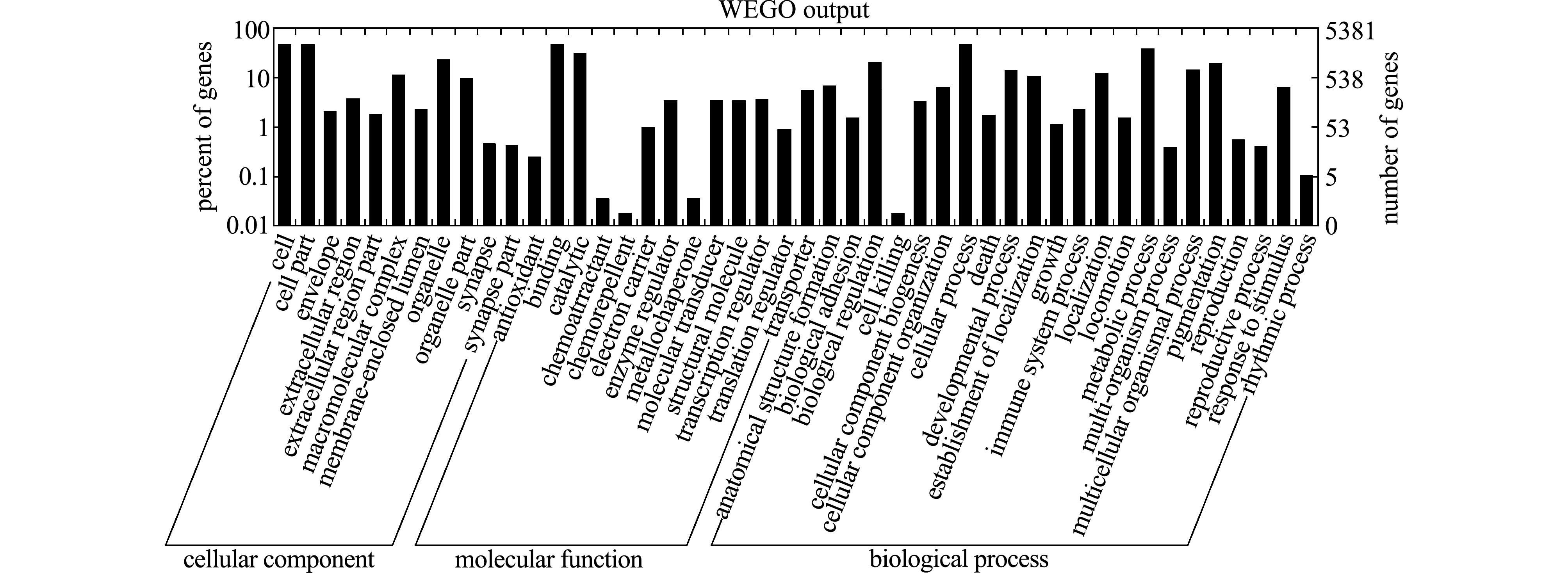
图2 鲮转录组的GO功能二级分类
Fig.2 Gene ontology (GO) (level 2) for transcriptome in mud carp Cirrhina molitorella
2.3微卫星预测
从2058条contigs中,总共鉴定出2438个微卫星。这些微卫星包括二碱基、三碱基、四碱基、五碱基和六碱基重复(阈值分别被设定为8、5、5、5、5)。以侧翼序列50 bp为阈值筛选,获得侧翼序列大于50 bp的微卫星共1379个,并为后续的PCR验证设计了相关的引物(表2)。
表2鲮转录组序列中微卫星的分布情况
Tab.2StatisticsofmicrosatellitesoftranscriptomefrommudcarpCirrhinamolitorella
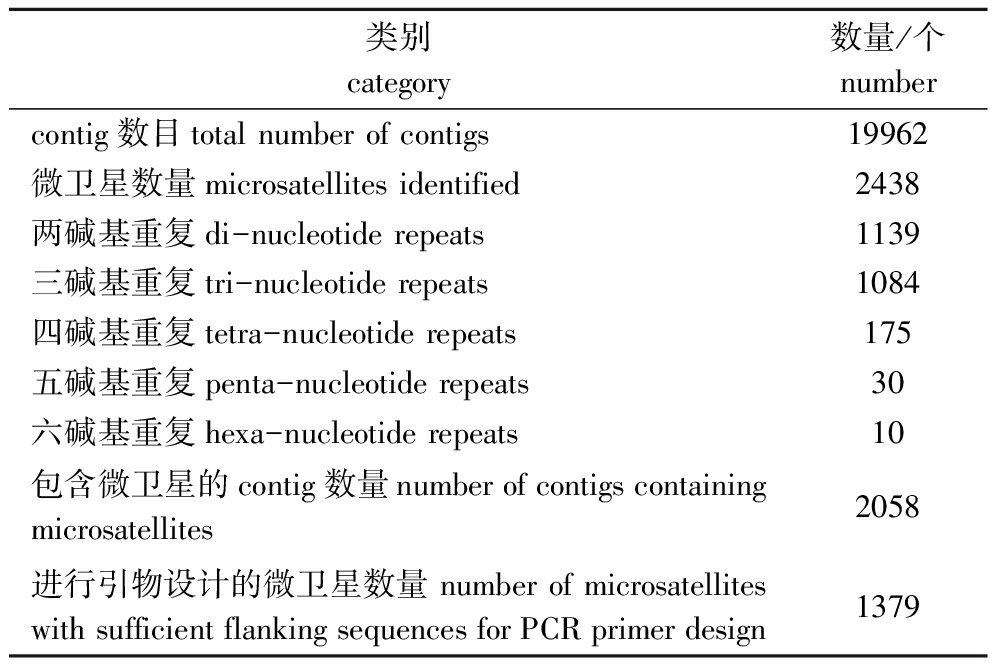
类别category数量/个numbercontig数目totalnumberofcontigs19962微卫星数量microsatellitesidentified2438两碱基重复di-nucleotiderepeats1139三碱基重复tri-nucleotiderepeats1084四碱基重复tetra-nucleotiderepeats175五碱基重复penta-nucleotiderepeats30六碱基重复hexa-nucleotiderepeats10包含微卫星的contig数量numberofcontigscontainingmicrosatellites2058进行引物设计的微卫星数量numberofmicrosatelliteswithsufficientflankingsequencesforPCRprimerdesign1379
2.4SNP分析
利用转录组作为参考序列,使用BWA和SAMtools程序对鲮的外显子区域进行SNP发掘,结果显示,总共得到5014个SNP位点,包括A-G、C-T、A-C、G-T、A-T和G-C六种类型的SNP。在所有类型的SNP中,A-G和C-T类型的比例最高,占所有SNP位点的72.6%,A-C、G-T、A-T和G-C这4种SNP类型有相似的比例,占总量的27.4%(表3)。
表3鲮转录组SNP位点的分类
Tab.3ClassificationofSNPsidentifiedtranscriptomefrommudcarpCirrhinamolitorella
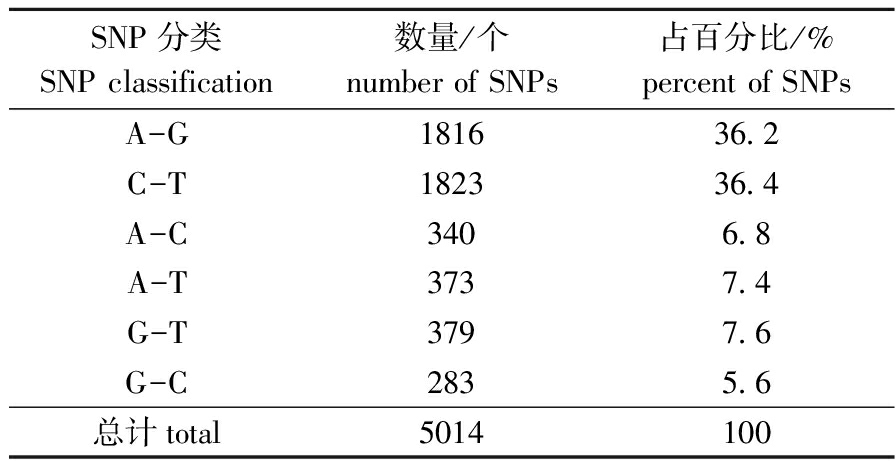
SNP分类SNPclassification数量/个numberofSNPs占百分比/%percentofSNPsA-G181636 2C-T182336 4A-C3406 8A-T3737 4G-T3797 6G-C2835 6总计total5014100
2.5全长cDNA的鉴定
根据Ensembl斑马鱼蛋白数据库,通过TargetIdentifier在线工具对所有的contigs进行完整ORF查找。结果共鉴定出5931个全长cDNA序列(e-value为1E-5)(表4),全长cDNA的长度分布如图3所示。
2.6鲮和鲤转录组的比较
在鲤科鱼类中,鲤的全鱼转录组已经被测定[4]。通过比较,鲮的总reads数要少于鲤,但平均reads长度比鲤长。鲤的contig条数也要比鲮多,鲮的N50为1509 bp,这表明Newbler软件用于转录组组装的分析结果比较理想。利用NCBI的非冗余蛋白库进行比对,结果显示,13 657条contigs得到注释,占总contigs数的68.4%,鲤为76.2%;此外,两者具有GO注释的特异蛋白的比例也基本一致,分别为50.9%和49.8%(表5)。
表4TargetIdentifier分析结果
Tab.4SummaryoftheTargetIdentifierreport

类别category数量number/个总序列totalnumberofsequences19962被注释的序列sequenceswithhit13534全长序列full-lengthsequences5931短的全长序列shortfull-lengthsequences11975′端部分序列5′sequencedpartialsequences26423′端部分序列3′sequencedpartialsequences3673全长并且ORF完整的序列full-length&ORFcompletelysequencedsequences5332
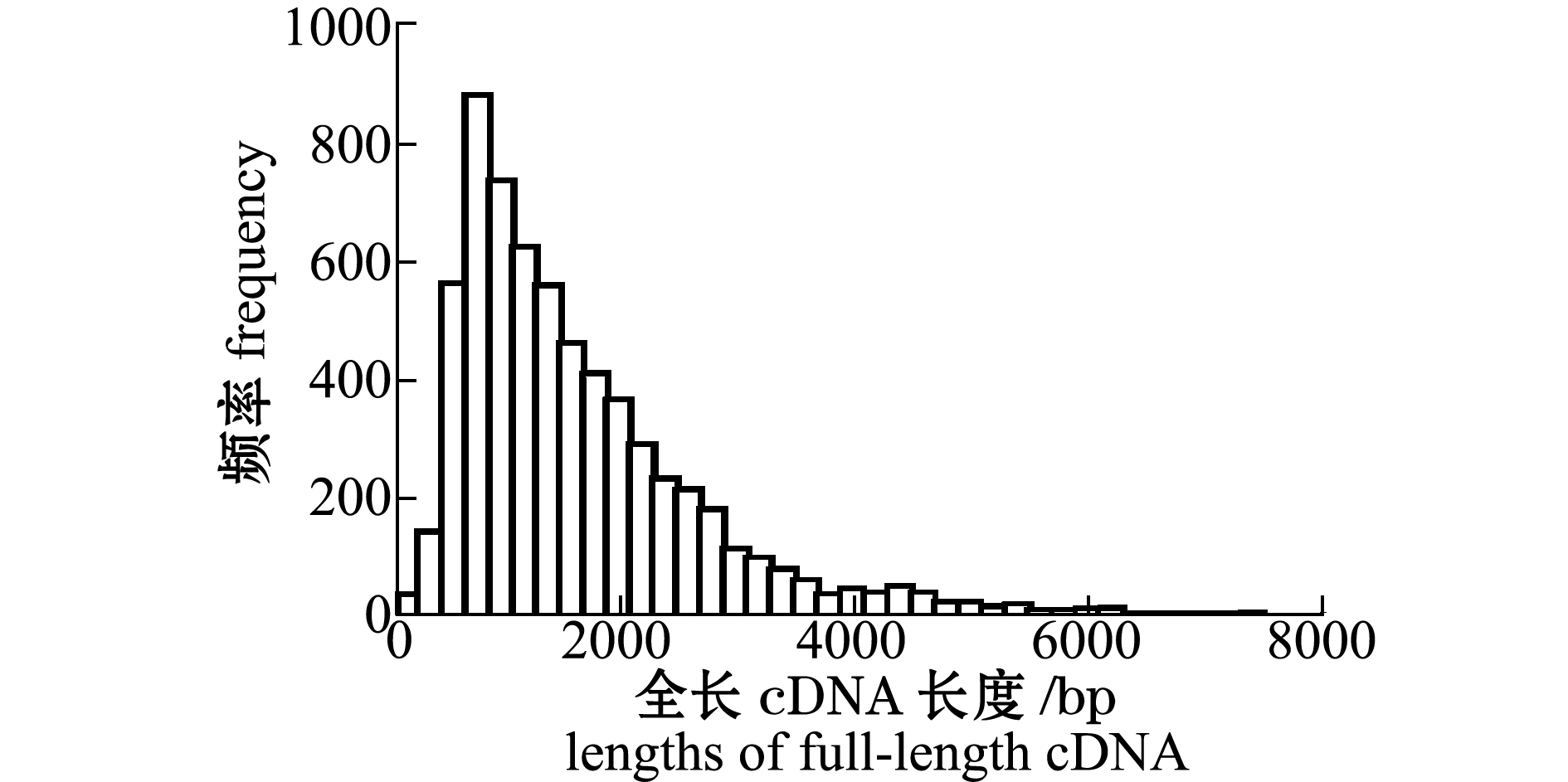
图3 鲮转录组全长cDNA的长度分布图
Fig.3 Length distributions of putative full-length cDNAs in mud carp transcriptome
2.7构建鲮转录组数据库
为了更便捷地应用鲮转录组的分析结果,根据转录组装配序列、基因注释结果、蛋白家族、SNP、微卫星等信息构建了鲮转录组数据库和网站(http://genomics.cafs.ac.cn/atd_www/)。通过该数据库,可以进行鲮基因的序列调取、序列比对、斑马鱼同源序列下载等,为鲮的基因克隆等基础遗传研究和分子育种提供了有力的支持。
目前,基于RNA-Seq的二代测序技术已经改变了转录组的研究方式[5-6],RNA测序在揭示转录组的复杂性、基因的鉴定、相关标记的分析、非编码RNA分析和可变剪切分析等方面起着重要的作用[7-9]。RNA测序技术在鱼类的研究中也发挥了重要作用。利用该技术在斑马鱼早期发育的不同阶段,鉴定出差异基因,为斑马鱼的发育提供了理论基础[10]。在进化研究方面,Zheng等[11]利用该技术,通过斑马鱼鳔的转录组同人肺的比较,发现了鱼类鳔和人肺是同源器官的分子生物学证据;Shin等[12]通过对3种南极鱼转录组的研究鉴定出一些抗寒基因;Liu等[7]对斑点叉尾鮰Ictalurusfurcatus升温至其失去平衡的试验中鉴定出鱼适应高温胁迫的相关基因及生理过程;草鱼经GCRV感染后,在头肾中鉴定出很多与免疫相关的基因和代谢途径,为培育出抗GCRV的品种提供了理论基础[13]。在性别差异方面,Sun等[14]利用该技术,通过比较斑点叉尾鮰雌雄性腺的基因表达差异,鉴定出影响性别的决定基因,为研究鱼的性别决定及分化提供了可靠的资料。另外,将RNA-Seq技术用于大量的SNP鉴定,为遗传育种、种群遗传学和资源评估等研究提供了丰富的标记资源,如应用于鲤C.carpioL.[15]、斑点叉尾鮰[9]、虹鳟Oncorhynchusmykiss[16]、红鳍东方鲀Takifugurubripes[17-18]等鱼类中。
表5鲮和鲤转录组的比较
Tab.5ComparisonoftranscriptomebetweenmudcarpCirrhinamolitorellaandcommoncarpCyprinuscarpioL.
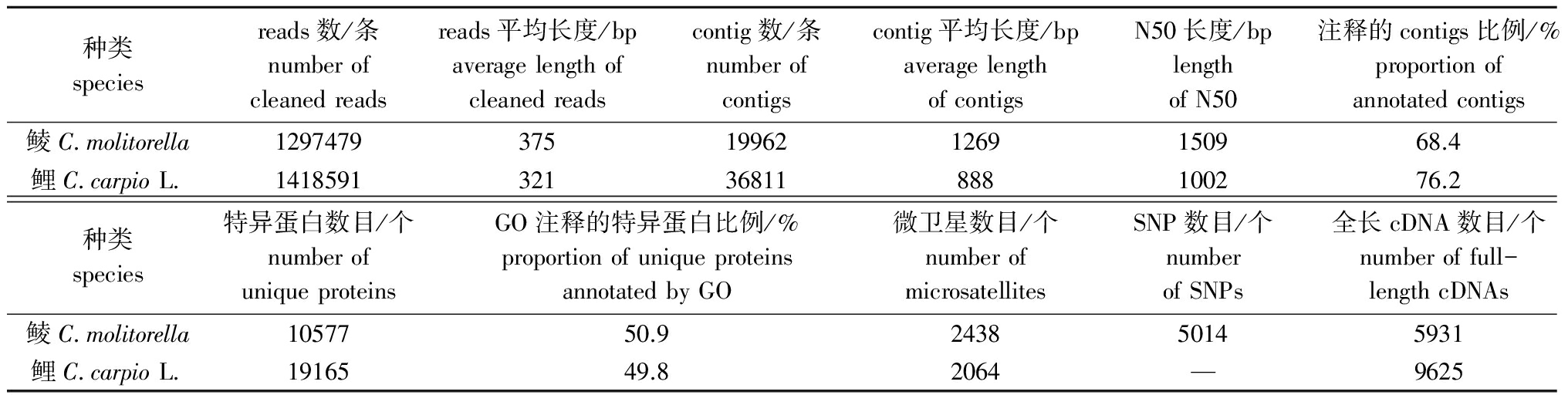
种类speciesreads数/条numberofcleanedreadsreads平均长度/bpaveragelengthofcleanedreadscontig数/条numberofcontigscontig平均长度/bpaveragelengthofcontigsN50长度/bplengthofN50注释的contigs比例/%proportionofannotatedcontigs鲮C molitorella1297479375199621269150968.4鲤C carpioL 141859132136811888100276.2种类species特异蛋白数目/个numberofuniqueproteinsGO注释的特异蛋白比例/%proportionofuniqueproteinsannotatedbyGO微卫星数目/个numberofmicrosatellitesSNP数目/个numberofSNPs全长cDNA数目/个numberoffull-lengthcDNAs鲮C molitorella1057750.9243850145931鲤C carpioL 1916549.82064—9625
本研究中,采用454测序技术对鲮进行转录组测序,分析鲮转录组的特征,共获得1 297 479条reads,组装后得到19 962条contigs,其中7314条contigs获得了GO的功能注释。另外,鉴定出5931个具有完整的ORF的全长cDNA序列,并且共鉴定出2438个微卫星和5014个SNP位点。以上结果均整合到了鲮转录组数据库中。近年来,对鲮的遗传和分子生物学方面的研究,初步阐述了鲮的遗传多样性[2],开展了少量的基因克隆与分子标记试验[1],并探讨了不同地理分布的鲮的进化关系以及与肌肉发育相关的基因。然而,在分子育种层面,由于基础数据资源的缺乏,尚无相关的研究进展。本研究中获得了鲮的转录组序列、基因注释结果和数千个分子标记,使该领域的研究人员可以大大加快研究的步伐,利用已有的家系样本,可对目前关心鲮的生长、抗寒等性状进行深入地分析。本研究中获得了鲮的转录组数据,并在此基础上对序列进行了注释,获得了大量微卫星及SNP信息,构建了鲮的数据库和网站,为深入开展鲮的生理生化机制研究、分子标记辅助的遗传育种、种群遗传学和资源评估等研究提供了丰富的标记资源。
参考文献:
[1] 钟茂春,郑光明,赵建,等.鲮Myf5基因克隆及其SNPs分析[J].中国水产科学,2010,17(4):681-688.
[2] 张丹丹,郑光明,朱新平,等.西江野生鲮与养殖群体的遗传分析[J].华南农业大学学报,2009,30(3):81-85.
[3] 刘佳瑶,赵建,郑光明,等.鲮微卫星DNA分子标记的筛选与遗传多样性分析[J].基因组学与应用生物学,2012,31(4):374-380.
[4] Ji P,Liu G,Xu J,et al.Characterization of common carp transcriptome:sequencing,de novo assembly,annotation and comparative genomics[J].PLoS One,2012,7(4):e35152.
[5] Wang Z,Gerstein M,Snyder M.RNA-Seq:a revolutionary tool for transcriptomics[J].Nat Rev Genet,2009,10:57-63.
[6] Anisimov S V.Serial analysis of gene expression (SAGE):13 years of application in research[J].Curr Pharm Biotechnol,2008,9:338-350.
[7] Liu S,Wang X,Sun F,et al.RNA-Seq reveals expression signatures of genes involved in oxygen transport,protein synthesis,folding and degradation in response to heat stress in catfish[J].Physiol Genomics,2013,45(12):462-476.
[8] Liu S,Zhang Y,Zhou Z,et al.Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote[J].BMC Genomics,2012,13:595.
[9] Liu S,Zhou Z,Lu J,et al.Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array[J].BMC Genomics,2011,12:53.
[10] Vesterlund L,Jiao H,Unneberg P,et al.The zebrafish transcriptome during early development[J].BMC Dev Biol,2011,11:30.
[11] Zheng W,Wang Z,Collins J E,et al.Comparative transcriptome analyses indicate molecular homology of zebrafish swim bladder and mammalian lung[J].PLoS One,2011,6(8):e24019.
[12] Shin S C,Kim S J,Lee J K,et al.Transcriptomics and comparative analysis of three Antarctic notothenioid fishes[J].PLoS One,2012,7(8):e43762.
[13] Chen J,Li C,Huang R,et al.Transcriptome analysis of head kidney in grass carp and discovery of immune-related genes[J].BMC Vet Res,2012,8:108.
[14] Sun F,Liu S,Gao X,et al.Male-biased genes in catfish as revealed by RNA-Seq analysis of the testis transcriptome[J].PLoS One,2013,8(7):e68452.
[15] Xu J,Ji P,Zhao Z,et al.Genome-wide SNP discovery from transcriptome of four common carp strains[J].PLoS One,2012,7(10):e48140.
[16] Salem M,Vallejo R L,Leeds T D,et al.RNA-Seq identifies SNP markers for growth traits in rainbow trout[J].PLoS One,2012,7(5):e36264.
[17] Cui J,Liu S,Zhang B,et al.Transciptome analysis of the gill and swim bladder ofTakifugurubripesby RNA-seq[J].PLoS One,2013,9(1):e85505.
[18] Cui J,Wang H,Liu S,et al.SNP discovery from transcriptome of the swim bladder ofTakifugurubripes[J].PLoS One,2013,9(3):e92502.
Abstract:The transcriptome of a mud carpCirrhinamolitorellawas sequenced using 454 sequencing technique, and the global transcriptome bioinformatics analysis was conducted using Newbler de novo assembly subsequently to understand genomic structure of mud carp. The results showed that the total reads were found to be 1 297 479 with the length of 486 586 191 bp. De novo assembly generated 19 962 contigs with the average length of 1269 bp, and the length of 1509 bp to N50.Based on Gene Ontology (GO) functional classifications, all unique transcripts were grouped into function categories, such as cellular component, molecular function, and biological process, and acquired many important genes on growth and development, which provides data for future gene functional studies. In addition, 5931 full-length cDNAs, 2438 microsatellite markers (SSR) and 5014 single nucleotide polymorphisms (SNPs) were identified. The findings were integrated into a database, providing with many gene sources which can be readily adopted for the design of marker assisted selection, population genetics and resource evaluation.
Key words:Cirrhinamolitorella; transcriptome; high-throughput RNA sequencing; de novo assembly
DOI:10.3969/J.ISSN.2095-1388.2014.06.003
文章编号:2095-1388(2014)06-0556-05
收稿日期:2014-03-25
基金项目:国家 “863”计划项目(2011AA100401);国家公益性行业(农业)科研专项(200903045);中央级公益性科研院所基本科研业务费资助项目(2013C011,2013C010)
中图分类号:Q954.4
文献标志码::A