仿刺参3个地理群体ITS1序列变异及系统发生分析
姬南京,常亚青,高银雪,赵冲,宋坚
(大连海洋大学 农业部北方海水增养殖重点实验室,辽宁 大连 116023)
摘要:利用核糖体DNA第一内转录间隔区(Internal transcribed spacer 1, ITS1)序列,对中国大连(CD)、朝鲜罗津(KN)和俄罗斯海参崴(RV)仿刺参Apostichopusjaponicus3个群体的遗传结构进行分析。结果表明:经PCR扩增、克隆测序,获得长度为517~519 bp、524 bp两种类型的ITS1核苷酸序列,平均G+C含量(64.5%)显著高于A+T含量(35.5%);在仿刺参30个个体61条序列中共检测到49个变异位点,多态位点比例为9.33%,其中有4个简约信息位点,共有40种基因型,群体共享基因型为2个;遗传多样性分析表明,3个群体的遗传多样性丰富;分子方差分析显示,88.65%的变异来自于群体内部,群体间遗传分化较弱或中度分化;根据群体间遗传距离进行的聚类分析发现,KN群体与RV群体的亲缘关系较近,CD群体与KN、RV群体的亲缘关系较远。
关键词:仿刺参;ITS1序列;遗传多样性;遗传分化
仿刺参Apostichopusjaponicus(Selenka)隶属于棘皮动物门、游移亚门、海参纲、楯手目、刺参科、仿刺参属,主要分布于中国、日本、俄罗斯和韩国沿海,在中国主要分布于辽宁、河北、山东等北方沿海,具有较高的营养和药用价值[1]。近年来,仿刺参养殖规模迅速扩大,由于长期近亲繁殖、高密度养殖等原因,导致了养殖仿刺参个体出现抗病力下降和生长缓慢等问题,影响了仿刺参养殖业的持续发展[2]。国家级仿刺参新品种“水院1号”的成功培育,证明了杂交育种是获得仿刺参优良品种的有效途径之一。通常情况下,杂交亲本间遗传距离越大,杂种优势就越明显;物种的遗传多样性越丰富,其对环境的适应能力就越强。因此,分析仿刺参不同地理群体间的进化关系和遗传多样性,是进行仿刺参种质资源管理和保护的前提之一,也是杂种优势预测工作的突破口。
真核生物核糖体RNA基因(18S rRNA、5.8S rRNA和28S rRNA)以串联重复方式组成一个转录单元(rDNA)。而核糖体内转录间隔区(Internal transcribed spacer,ITS)是rDNA内部的间隔区域,最终不加入成熟的核糖体中,因此,受到的选择压力较小,进化速度快,很短的序列就能够提供比较丰富的遗传信息[3],其在海洋动物遗传多样性、系统进化等方面具有广泛的应用性[4-6]。
近年来,关于仿刺参群体遗传多样性研究主要采用同工酶[7]、ISSR[8]、微卫星[9]等技术,但利用ITS1序列对其进行遗传学分析的研究尚未见报道。本研究中,利用ITS1的多态性,对仿刺参3个地理群体进行遗传结构分析,以期从基因组水平上了解仿刺参资源的遗传背景,为仿刺参的杂交育种和种群资源管理提供参考依据。
1材料与方法
1.1材料
试验用仿刺参样品分别采自中国大连(CD)、朝鲜罗津(KN)、俄罗斯海参崴(RV),每个群体10个样本。
1.2方法
1.2.1 基因组DNA的提取 取活体仿刺参的管足6~7根,采用TIANamp Marine Animals DNA Kit试剂盒,提取仿刺参基因组总DNA,经10 g/L琼脂糖凝胶电泳检测后,作为PCR模板。
1.2.2 ITS1序列的扩增及克隆测序 根据GenBank中仿刺参(AB593815)的18 S和5.8 S保守区设计特异性引物,扩增ITS1全序列。扩增引物序列为
ITS1-F:5′ AAAGCCGTCACAGAGAGACA 3′,
ITS1-R:5′ CATCTAACTGCGTTCTTCATC 3′。
PCR体系(共50 μL):模板DNA 50~100 ng,25 μmol/L上下游引物各1 μL,10×Buffer 5 μL,25 mmol/L Mg2+4 μL,2.5 mmol/L dNTP 4 μL,5 U/μL Taq酶(TaKaRa公司)0.4 μL,用去离子水补至50 μL。PCR反应条件:94 ℃下预变性5 min;94 ℃下变性40 s,58 ℃下退火40 s,72 ℃下延伸1 min,共进行30个循环;最后在72 ℃下再延伸10 min。用10 g/L琼脂糖凝胶电泳检测,采用UNIQ-10柱式DNA胶回收试剂盒(上海生工有限公司)回收目的条带,与pMD-19T载体(TaKaRa公司)连接后转化到DH5α感受态细胞中,采用蓝白斑挑选阳性克隆,每个个体挑选3个阳性克隆子,委托上海英潍捷基生物技术有限公司进行测序。
1.3数据处理
利用DNAStar软件包进行序列编辑和校正,用Clustal X(1.83)软件进行多重比较,确定序列的边界长度。采用DnaSp 5.0软件统计单倍型数、单倍型多样性、平均核苷酸差异数、核苷酸多样性指数。采用Arlequin 3.0软件[10]中的分子变异分析(AMOVA)统计遗传变异来源及群体间分化程度。采用Mega 4.0软件[11]进行核苷酸组成分析,统计变异位点数、转换/颠换,利用Kitmura双参数模型计算群体间遗传距离,再根据群体间的遗传距离,采用邻接法(NJ)构建系统进化树。
2结果
2.1ITS1序列特征
PCR产物经克隆测序后,去掉两侧的18 S(33 bp)、5.8 S(57 bp)序列,经Mega 4.0软件分析表明,ITS1基因长度分成A、B两种类型:A型为517~519 bp,B型为524 bp。大部分序列为A型,而B型由6碱基(TCGAAG)插入造成,仅存在于KN群体的少数个体中。碱基组成分析表明,ITS1的平均G+C含量(64.5%)显著高于A+T含量(35.5%),3个群体之间的G+C含量没有差异。在525个比对位点中检测到49个变异位点,包括4个简约信息位点,多态位点比例为9.33%,转换/颠换值为2.94。
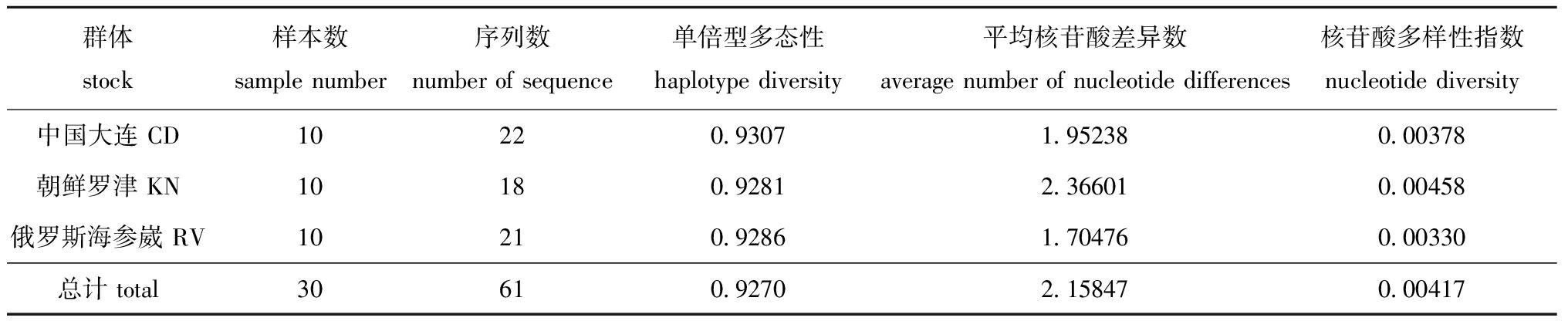
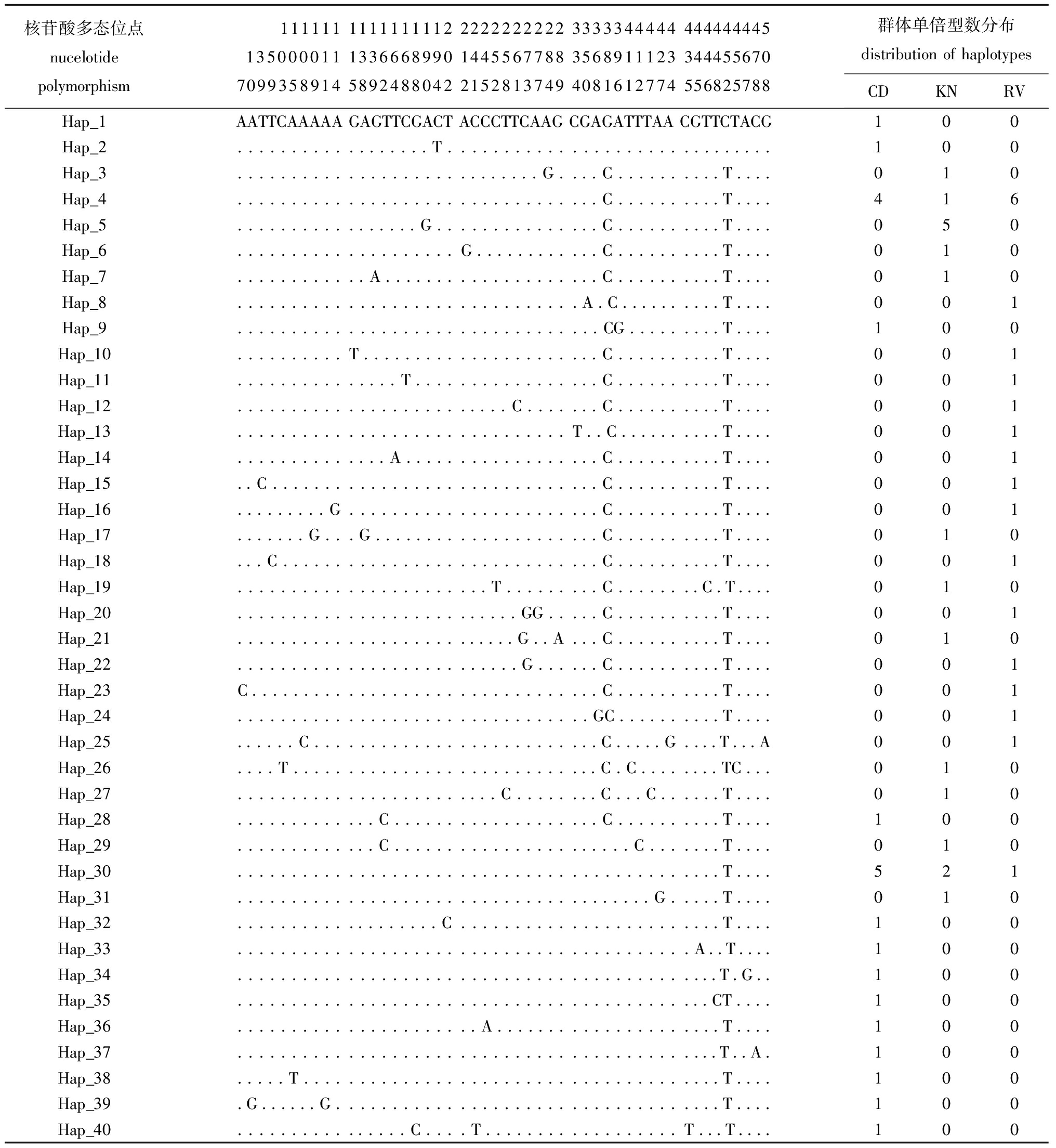
用DnaSp 5.0软件计算群体遗传多样性参数(表1),在3个群体61条序列中,共检出40种基因型,基因型变异位点如表2所示。从表2可见,基因型4和30为群体共享,其余基因型为群体所特有。
表1仿刺参3个群体ITS1序列遗传多样性参数
Tab.1GeneticdiversityparametersofITS1sequencesinthe3populationsofseacucumberApostichopusjaponicus
群体stock样本数samplenumber序列数numberofsequence单倍型多态性haplotypediversity平均核苷酸差异数averagenumberofnucleotidedifferences核苷酸多样性指数nucleotidediversity中国大连CD10220 93071 952380 00378朝鲜罗津KN10180 92812 366010 00458俄罗斯海参崴RV10210 92861 704760 00330总计total30610 92702 158470 00417
2.2基于ITS1群体的遗传变异
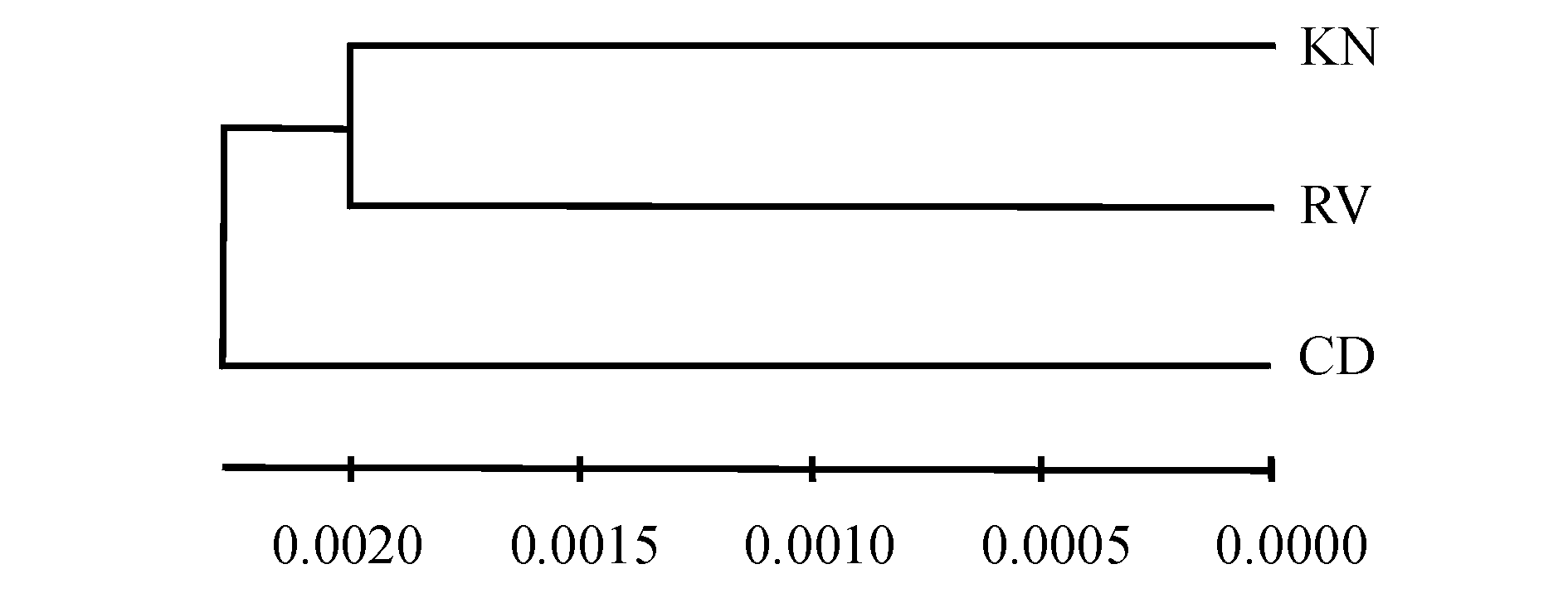
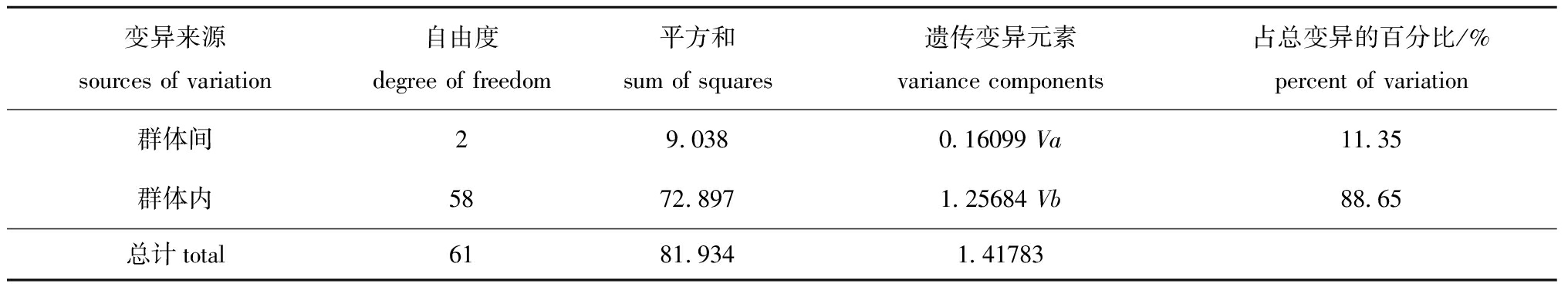
将每个群体作为一组,利用Arlequin 3.0软件进行AMOVA分析,估算3个群体间的遗传变异结构及变异来源,具体结果如表3所示。AMOVA分析结果显示,群体间的分子变异显著(FST=0.1135,P<0.05),表明种群间的变异占总变异的11.35%,88.65%的变异来自于群体内部。仿刺参3个群体的遗传分化系数见表4,CD群体与RV群体之间遗传分化系数最高,为0.192 7,其次为CD群体与KN群体之间的遗传分化系数,为0.109 6,RV群体与KN群体之间的遗传分化系数最低,为0.042 3。基于ITS1基因遗传距离构建3个群体的NJ聚类树,结果如图1所示。
表2 ITS1序列多态点及各单倍型在群体中的分布
Tab.2PolymorphicsitesofITS1sequencesandhaplotypeprofilesinthepopulations
核苷酸多态位点nucelotidepolymorphism1111111111111112222222222233333444444444444451350000111336668990144556778835689111233444556707099358914589248804221528137494081612774556825788群体单倍型数分布distributionofhaplotypesCDKNRVHap_1AATTCAAAAAGAGTTCGACTACCCTTCAAGCGAGATTTAACGTTCTACG100Hap_2..................T..............................100Hap_3............................G....C..........T....010Hap_4.................................C..........T....416Hap_5.................G...............C..........T....050Hap_6....................G............C..........T....010Hap_7............A....................C..........T....010Hap_8...............................A.C..........T....001Hap_9.................................CG.........T....100Hap_10..........T......................C..........T....001Hap_11...............T.................C..........T....001Hap_12.........................C.......C..........T....001Hap_13..............................T..C..........T....001Hap_14..............A..................C..........T....001Hap_15..C..............................C..........T....001Hap_16.........G.......................C..........T....001Hap_17.......G...G.....................C..........T....010Hap_18...C.............................C..........T....001Hap_19.......................T.........C........C.T....010Hap_20..........................GG.....C..........T....001Hap_21..........................G..A...C..........T....010Hap_22..........................G......C..........T....001Hap_23C................................C..........T....001Hap_24................................GC..........T....001Hap_25......C..........................C.....G....T...A001Hap_26....T............................C.C........TC...010Hap_27........................C........C...C......T....010Hap_28.............C...................C..........T....100Hap_29.............C......................C.......T....010Hap_30............................................T....521Hap_31......................................G.....T....010Hap_32...................C........................T....100Hap_33.........................................A..T....100Hap_34............................................T.G..100Hap_35...........................................CT....100Hap_36......................A.....................T....100Hap_37............................................T..A.100Hap_38.....T......................................T....100Hap_39.G......G...................................T....100Hap_40................C....T..................T...T....100
表3仿刺参群体遗传变异的分子方差分析(AMOVA)
Tab.3MolecularAMOVAanalysisofgeneticvariationinthepopulationsofseacucumberApostichopusjaponicus
变异来源sourcesofvariation自由度degreeoffreedom平方和sumofsquares遗传变异元素variancecomponents占总变异的百分比/%percentofvariation群体间29 0380 16099Va11 35群体内5872 8971 25684Vb88 65总计total6181 9341 41783
表4群体间的相对遗传距离(右上)和遗传分化系数(左下)
Tab.4Geneticdistanceofeachpopulation(upperright)andFSTamongthepopulations(lowerleft)

群体stock中国大连CD朝鲜罗津KN俄罗斯海参崴RV中国大连CD0 00470 0044朝鲜罗津KN0 10960 0040俄罗斯海参崴RV0 19270 0423
3讨论
3.1ITS1序列特征与遗传多样性分析
一般研究认为,如果一个基因的转换/颠换值小于2.0,则认为其突变已达到饱和,易受进化噪音的影响,在系统进化分析时需进行加权分析[12]。本研究中,ITS1序列转换/颠换值为2.94,表明ITS1序列的位点突变未达到饱和,可以用作仿刺参系统进化分析。另外,本研究中对仿刺参每个个体都进行了多克隆测序,发现其ITS1序列在个体内存在多态,造成有的个体内存在3种基因型。因此,笔者建议今后对仿刺参ITS1序列进行分析时,应考虑对单个个体进行多克隆测序。
遗传多样性是生物多样性的基础,指种内不同种群间或一个种群内部不同个体间的遗传变异,是生物进化和适应的基础,一个物种的遗传多样性越丰富,该物种对环境的适应能力就越强。本研究中,仿刺参3个群体核苷酸多样性指数为0.004 17,大于海蜇Rhopilemaesculentum野生群体(0.002 59)[13]、硬壳蛤Mercenariamercenaria养殖群体(0.003 02)[14],说明本研究中的仿刺参3个群体遗传多样性较为丰富。
3.2群体遗传分化与系统发生分析
根据群体遗传学理论,FST值可以表示群体间的分化程度,FST值为0~0.05时表示分化较弱,FST为0.05~0.15时表示遗传分化中等,FST为0.25以上时表示遗传分化极大[15]。本研究中,CD群体与RV、KN群体之间中度遗传分化,RV群体与KN群体之间分化较弱。Uthicke等[16]采用同工酶技术对新喀里多尼亚沿岸的糙海参进行遗传分析,发现距离150 km的地理群体之间形成基因流,遗传分化也不显著。有研究者认为,形成这一现象的主要原因是海洋无脊椎动物在幼虫时期长时间处于浮游状态,群体之间形成基因流,造成地理距离较远的群体之间遗传分化较弱[17]。
本研究中,基于ITS1序列分析仿刺参3个群体间的遗传距离为0.004 0~0.004 7,其中CD群体与其他两个群体之间遗传距离最远,RV群体与KN群体遗传距离最近。Yao等[8]采用ISSR技术分析青岛与蓬莱仿刺参之间的遗传距离为0.079 6;Sun等[18]采用线粒体DNA标记技术分析体色不同的仿刺参之间遗传距离为0.007 2~0.010 0。笔者认为,造成遗传距离明显差异的原因主要是由不同的分子标记和不同的地理群体造成的。牛东红等[19]曾对缢蛏8个群体分别采用微卫星和COⅠ标记进行分析,群体间遗传距离分别为0.078 5~0.275 7、0.005 9~0.011 1。本研究中,根据群体间的遗传距离,构建的NJ树发现,RV群体与KN群体聚为一支,CD群体为独立一支,聚类结果与它们的地理位置分布一致,聚类关系符合地理隔离模式,这种现象在海洋动物中已多次被发现[20-21]。
综上所述,本研究中的仿刺参3个群体具有较高的遗传多样性,种质资源丰富。但长期人工育苗增加了个体间的基因交流,可引起群体遗传多样性降低。因此,为了仿刺参资源的可持续发展,应加强仿刺参的遗传多样性检测。
参考文献:
[1] 常亚青,丁君,宋坚,等.海参、海胆生物学研究与养殖[M].北京:海洋出版社,2004:3-4.
[2] 谭杰,孙慧玲,刘萍,等.仿刺参自然群体和养殖群体间遗传变异的微卫星标记研究[J].海洋水产研究,2007,28(3):38-43.
[3] Hillis D M,Dixon M T.Ribosomal DNA:molecular evolution and phylogenetic inference[J].Q Rev Biol,1991,66(4):411-453.
[4] 孟学平,高如承,申欣,等.西施舌5个地理群体ITS1 序列变异及系统发生分析[J].生态学报,2011,30(20):5555-5561.
[5] 马朋,刘萍,李健.脊尾白虾3个野生群体 ITS1 序列分析及其亲缘关系分析[J].水产学报,2012,36(8):1185-1192.
[6] 李远宁,马朋,刘萍,等.三疣梭子蟹 (Portunustrituberculatus)4个野生群体ITS1序列分析及系统进化分析[J].海洋与湖沼,2012,43(4):768-774.
[7] 高悦勉,孙静波.刺参种群同工酶的生化遗传分析[J].大连水产学院学报,2004,19(1):30-34.
[8] Yao B,Hu X L,Bao Z M,et al.Genetic variation in two sea cucumber (Apostichopusjaponicus) stocks revealed by ISSR markers[J].Chin J Oceanol Limnol,2007,25(1):91-96.
[9] Chang Y Q,Feng Z G,Yu J P,et al.Genetic variability analysis in five populations of the sea cucumberStichopus(Apostichopus)japonicusfrom China,Russia,South Korea and Japan as revealed by microsatellite markers[J].Mar Ecol,2009,30(4):455-461.
[10] Excoffier L,Laval G,Schneider S.Arlequin ver 3.0:an integrated software package for population genetics data analysis[J].Evol Bioinform Online,2005(1):47-50.
[11] Tamura K,Dudley J,Nei M,et al.MEGA4:molecular evolutionary genetics analysis (MEGA) software version 4.0[J].Mol Biol Evol,2007,24(8):1596-1599.
[12] Knight A,Mindell D P.Substitution bias,weighting of DNA sequence evolution,and the phylogenetic position of Fea’s viper[J].Syst Biol,1993,42(1):18-31.
[13] 孙国华,刘相全,杨建敏,等.海蜇养殖群体及自然捕获群体ITS序列遗传分析[J].海洋科学,2010,34(10):90-95.
[14] 吴琪,潘鹤婷,潘宝平.帘蛤科两种经济贝类种群的ITS-1序列遗传多样性分析[J].天津师范大学学报,2007,27(1):20-23.
[15] 袁媛,高玮玮,吴琪,等.黄、渤海地区青蛤(Cyclinasinensis)种群的ITS序列遗传变异与遗传结构分析[J].海洋与湖沼,2008,39(6):665-670.
[16] Uthicke S,Purcell S.Preservation of genetic diversity in restocking of the sea cucumberHolothuriascabrainvestigated by allozyme electrophoresis[J].Can J Fish Aquat Sci,2004,61(4):519-528.
[17] 谭杰,孙慧玲,刘萍,等.3个仿刺参地理种群遗传变异的微卫星DNA分析[J].水产学报,2007,31(4):437-442.
[18] Sun X J,Li Q,Kong L F.Comparative mitochondrial genomics within sea cucumber (Apostichopusjaponicus):provide new insights into relationships among color variants[J].Aquaculture,2010,309(1/4):280-285.
[19] 牛东红,冯冰冰,刘达博,等.浙闽沿海缢蛏群体遗传结构的微卫星和线粒体COI序列分析[J].水产学报,2012,35(12):1805-1813.
[20] 吕振明,李焕,吴常文,等.中国沿海六个地理群体短蛸的遗传变异研究[J].海洋学报,2010,32(1):130-138.
[21] Arnaud S,Monteforte M,Galtier N,et al.Population structure and genetic variability of pearl oysterPinctadamazatlanicaalong Pacific coasts from Mexico to Panama[J].Conservation Genetics,2000,1(4):299-307.
VariationinITS1sequencesandphylogeneticanalysisofthreegeographicalstocksofseacucumberApostichopusjaponicus
JI Nan-jing,CHANG Ya-qing,GAO Yin-xue,ZHAO Chong,SONG Jian
(Key Laboratory of Mariculture & Stock Enhancement in North China’s Sea, Ministry of Agriculture, Dalian Ocean University, Dalian 116023, China)
Abstract:The genetic diversity and genetic differentiation were studied in three wild populations of sea cucumberApostichopusjaponicusfrom Dalian China (CD),Rajin North Korea (KN) and Vladivostok Russia(RV) by internal transcribed spacer 1(ITS1). Two different length of ITS1 sequence (517-519 bp and 524 bp) were obtained via cloning and sequencing of PCR products, with higher mean contents of G+C(64.5%) than those of A+T(35.5%). There were 49 variation sites from 61 sequences in the 3 populations, with polymorphic sites of 9.33%, and 4 parisimony informative among 40 genotypes including 2 shared genotypes. Genetic diversity parameters showed high genetic diversity among populations. The fixation indices (FST) of analysis of molecular variance (AMOVA) within populations was found to be 88.65%,indicating that the three populations had weak or only moderately genetic differentiation. The molecular phylogenetic tree constructed with NJ method based on genetic distance revealed that there was closer linkage between KN population and RV population than between them and CD population.
Key words:Apostichopusjaponicus; ITS1 sequences; genetic diversity; genetic differentiation
DOI:10.3969/J.ISSN.2095-1388.2014.05.005
文章编号:2095-1388(2014)05-0449-05
收稿日期:2013-12-15
基金项目:国家自然科学基金资助项目(31072230);国家“863”计划项目(2012AA10A412)
作者简介:姬南京(1988—), 男, 硕士研究生。E-mail:jinanjing@126.com
通信作者:常亚青(1967—), 男, 博士生导师。E-mail:yqchang@dlou.edu.cn
中图分类号:Q347;S917.4
文献标志码::A