中国帘蛤目16种经济贝类DNA条形码及分子系统发育的研究
王琳楠1,闫喜武1,秦艳杰1,聂鸿涛1,牛泓博1,张国范2
(1.大连海洋大学水产与生命学院辽宁省贝类良种繁育工程技术研究中心,辽宁大连116023;2.中国科学院海洋研究所海洋生物技术研发中心,山东青岛266071)
摘要:对中国帘蛤目主要经济贝类的分子系统发育进行了研究,应用DNA条形码通用引物扩增了6种中国近海帘蛤目经济贝类共计60个个体的COI基因片段,与GenBank收录的10种帘蛤目贝类50条同源序列进行比对。结果表明:帘蛤目贝类COI基因存在碱基插入和缺失现象,在16个物种中有6个物种存在103个插入和缺失位点,其中杂色蛤仔Ruditapes variegata、裂纹哥特蛤Katelysia hiantina插入和缺失位点均为30个,大竹蛏Solen grandis为27个;碱基的组成出现偏倚现象,A+T含量 (64.2%)明显高于G+C含量(35.8%);基于K2P模型的计算,16个物种的种内平均遗传距离为0.010 6,种间平均遗传距离为0.388 4,后者是前者的21.34倍;系统发育树聚类分析表明,COI基因在科、属、种水平上的鉴定及其系统进化关系重构方面与传统的形态学分类一致性较高。研究表明,线粒体COI基因作为帘蛤目贝类DNA条形码在物种鉴定的适用性上提供了一定的依据,同时也为形态分类系统提供了必要补充。
关键词:帘蛤目;COI基因;系统发育;分类鉴定;DNA条形码
帘蛤目Veneroida隶属于软体动物门Mollusca、双壳纲Bivalvia,是世界上分布较广的贝类[1]。中国的帘蛤目贝类种类丰富,已有记录的近千种,若仅仅依据壳型、放射肋、小月面等外部形态特征进行种属水平的形态学分类难度很大[2-3],需要以基于分子生物学的分析方法进行补充。DNA条形码技术 (DNA barcoding)是利用COI基因构建物种的鉴别体系[4-5],现已成为物种鉴定的一门新兴技术。DNA条形码的两大主要目的是鉴定已知种和发现新种,它相对于传统的生物鉴定的优势就是可以揭示隐存种。近年来,COI基因序列已经广泛应用于贝类的种质鉴定[6-10]、 种群遗传结构分析[11-13]、分子系统发生研究[14-21]等领域。 研究证实,COI基因序列适用种内、种间和属以上阶元的遗传多样性分析[22-25]。本研究中以中国黄渤海、东海、南海16种重要的帘蛤目经济贝类为材料,由于传统的形态学鉴定一般都是根据壳形、铰合部和闭壳肌痕等形态特征来进行鉴定,而实际上壳形的多变以及发育中存在的中间形态和中间生境,使得形态学分类比较困难,为此本研究基于COI序列的DNA条形码技术对这些贝类进行分析,从而证明DNA条形码在帘蛤目贝类辅助物种鉴定中的适用性,旨在为全球贝类DNA条形码数据库提供可靠的资料。
1 材料与方法
1.1 材料
试验用60个帘蛤目样品均采自中国沿海地区,参照 《中国海产双壳类图志》进行形态学鉴定,这些样品隶属于4科、5属、6种,分别为樱蛤科亮樱蛤属1种、竹蛏科竹蛏属2种、帘蛤科畸心蛤属1种、帘蛤科镜蛤属1种、绿螂科绿螂属1种。
1.2 方法
1.2.1 DNA的提取 从GenBank 中筛选出已经发表的帘蛤目COI基因序列信息50条,分别属于帘蛤目3科、9属、10种,共选取帘蛤目16个种, 110个个体进行研究。选取长牡蛎为外类群 (表 1)。试验时每个种取10~12个,取其斧足、闭壳肌或者外套膜约50 mg,采用酚氯仿法抽提DNA。DNA溶于TE中,于-20℃下保存备用。
表1 16种帘蛤目贝类的COI基因信息
Tab.1 Information of COI genes in 16 species in Veneroida

组别group物种species分类地位taxonomy样品数sample numbers GenBank登录号GenBank accession No. GP1 四角蛤蜊Mactra veneriformis帘蛤目蛤蜊科蛤蜊属5 GQ864246~GQ864250 GP2 虹光亮樱蛤Moerella iridescens帘蛤目樱蛤科亮樱蛤属 10 JN645202~JN645205,JN645207,JN645208, KC137303~KC137306 GP3 缢蛏Sinonovacula constrzcta帘蛤目截蛏科缢蛏属 5 FJ555180~FJ555184 GP4 大竹蛏Solen grandis帘蛤目竹蛏科竹蛏属 10 AB064983~AB064985,JN645206, KC137307~KC137312 GP5 长竹蛏S.strictus帘蛤目竹蛏科竹蛏属 10 JN645218~JN645220, KC137313~KC137319 GP6 畸心蛤Anomalocadia producta帘蛤目帘蛤科畸心蛤属 10 JN593283,JN645210,JN645211, KC137320~KC137326 GP7 江户布目蛤Protothaca jedoensis帘蛤目帘蛤科布目蛤属 5 HQ703066~HQ703070 GP8 日本镜蛤Dosinia japonica帘蛤目帘蛤科镜蛤属 10 HQ703138,HQ703137,GQ8552811, HM124575,JN645216,JN645217, KC137327~KC137330 GP9 缀锦蛤Tapes literatus帘蛤目帘蛤科缀锦蛤属 5 HQ703219~HQ703223 GP10 菲律宾蛤仔Ruditapes philippinarum帘蛤目帘蛤科帘蛤属 5 JN054628~JN054632 GP11 杂色蛤仔R.variegata帘蛤目帘蛤科帘蛤属 5 HQ703313~HQ703317 GP12 巴非蛤Paphia papilionacea帘蛤目帘蛤科巴非蛤属 5 HQ703239~HQ703243 GP13 裂纹哥特蛤Katelysia hiantina帘蛤目帘蛤科哥特蛤属 5 HQ703291~HQ703295 GP14 文蛤Meretrixmeretrix帘蛤目帘蛤科文蛤属 5 HQ703159~HQ703163 GP15 青蛤Cyclina sinensis帘蛤目帘蛤科青蛤属 5 HQ703127~HQ703131 GP16 中国绿螂Glauconome chinensis帘蛤目绿螂科绿螂属 10 JN645212~JN645215, KC137331~KC13736 OUT 长牡蛎Cassostrea gigas珍珠目牡蛎科牡蛎属1 AF280608
1.2.2 PCR扩增及产物测序 COI片段采用无脊椎动物通用的COI引物序列LCO1490(5'GGTCAACAAATCATAAAGATATTGG 3')和 HC02198 (5'TAAACTTCAGGGTGACCAAAAAATCA 3')[26]。引物由英潍捷基 (上海)贸易有限公司合成。扩增产物经20 g/L琼脂糖凝胶电泳检测后,由Invitrogen(上海)有限公司进行双向测序。
1.3 数据处理
利用 Clustal X 1.81软件进行序列比对排序[27],经DnaSP 4.0[28]单倍型分析后,将单倍型序列提交GenBank数据库。利用Mega 4.0软件统计不同样本的核苷酸组成、突变位点、遗传距离、转换/颠换值[18,29];利用DnaSP 4.0计算单倍型数量、单倍型多态性、核苷酸多样性指数、多态性位点数,以及插入和缺失位点数;使用Mega 4.0中的Kimura双参数法(K2P)计算种内和种间的遗传距离,并构建基于K2P的邻接树(neighbor joining, NJ)和最大简约树(maximum parsimony,MP)[30]。
2 结果
2.1 帘蛤目COI序列分析
本研究中,对6种帘蛤目的COI基因进行PCR扩增,挑选较好的PCR产物进行测序,经过比对和校正后提交GenBank数据库,得到的登录号见表1。将研究中测定及下载的所有COI序列进行整理,并裁剪为同一长度628 bp进行分析。统计结果表明,在研究的110个个体中共产生了56个单倍型,单倍型多态性指数为0.533 0~1.000 0;核苷酸多态性指数为0.001 07~0.088 04;在16个物种 (GP1~GP16)中有6个物种存在碱基插入和缺失现象,占到了37.50%,共存在103个插入和缺失位点 (表2)。
表2 16种帘蛤目贝类的COI基因序列特征
Tab.2 The characters of COI sequences from 16 species in Veneroida
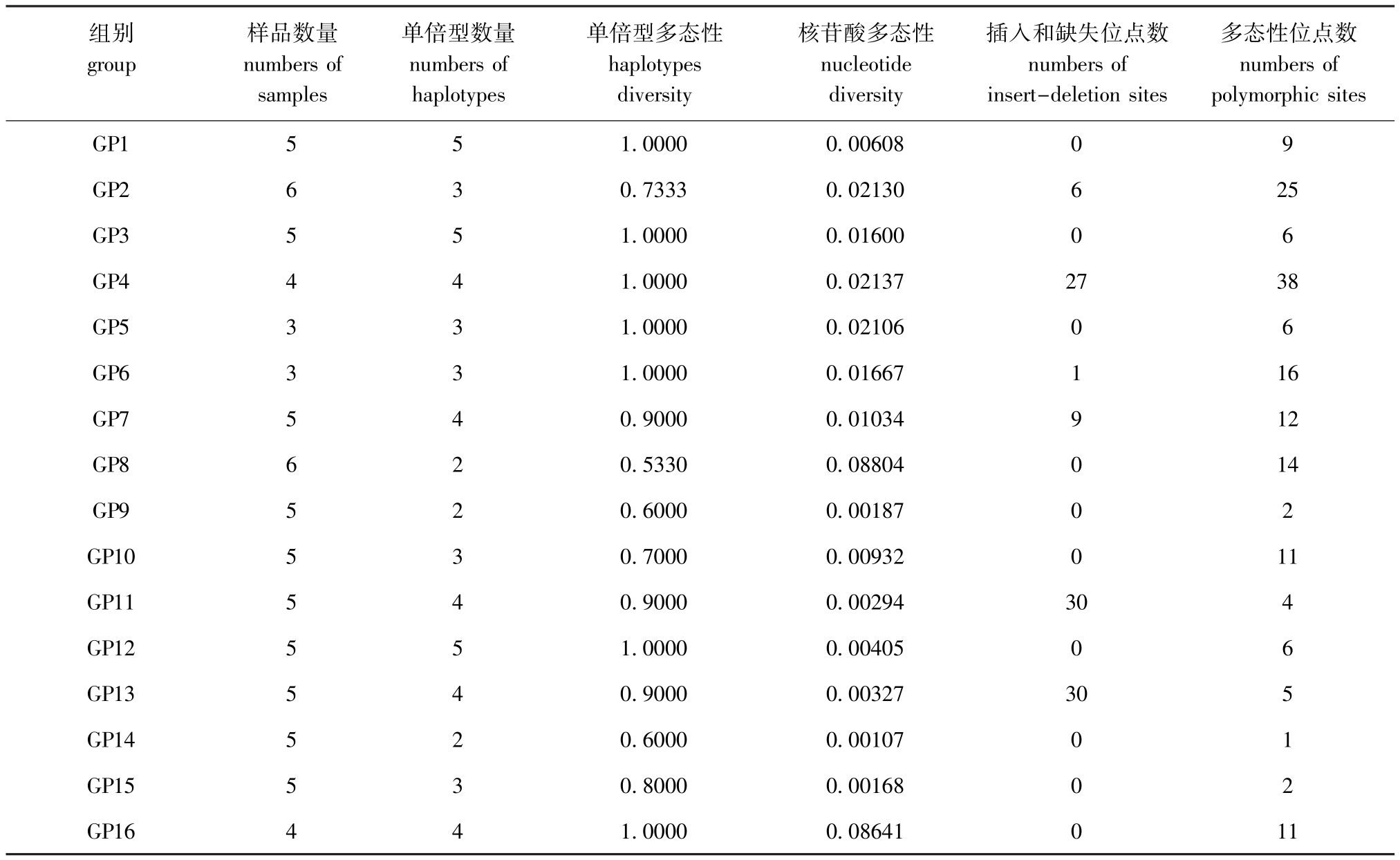
组别group样品数量numbers of samples单倍型数量numbers of haplotypes单倍型多态性haplotypes diversity核苷酸多态性nucleotide diversity插入和缺失位点数numbers of insert-deletion sites多态性位点数numbers of polymorphic sites GP1 5 5 1.0000 0.00608 0 9 GP2 6 3 0.7333 0.02130 6 25 GP3 5 5 1.0000 0.01600 0 6 GP4 4 4 1.0000 0.02137 27 38 GP5 3 3 1.0000 0.02106 0 6 GP6 3 3 1.0000 0.01667 1 16 GP7 5 4 0.9000 0.01034 9 12 GP8 6 2 0.5330 0.08804 0 14 GP9 5 2 0.6000 0.00187 0 2 GP10 5 3 0.7000 0.00932 0 11 GP11 5 4 0.9000 0.00294 30 4 GP12 5 5 1.0000 0.00405 0 6 GP13 5 4 0.9000 0.00327 30 5 GP14 5 2 0.6000 0.00107 0 1 GP15 5 3 0.8000 0.00168 0 2 GP16 4 4 1.0000 0.08641 0 11
110条帘蛤目贝类COI序列的平均碱基组成: T为 41.4%,C为 14.3%,A为 22.8%,G为21.4%,其中A+T的含量 (64.2%)明显高于G+ C的含量 (35.8%),碱基出现明显的偏倚性,符合线粒体碱基组成的特点 (表3)。
表3 16种帘蛤目贝类的COI序列碱基组成的频率
Tab.3 Average nucleotide frequencies of COI sequences in 16 species in Veneroida %
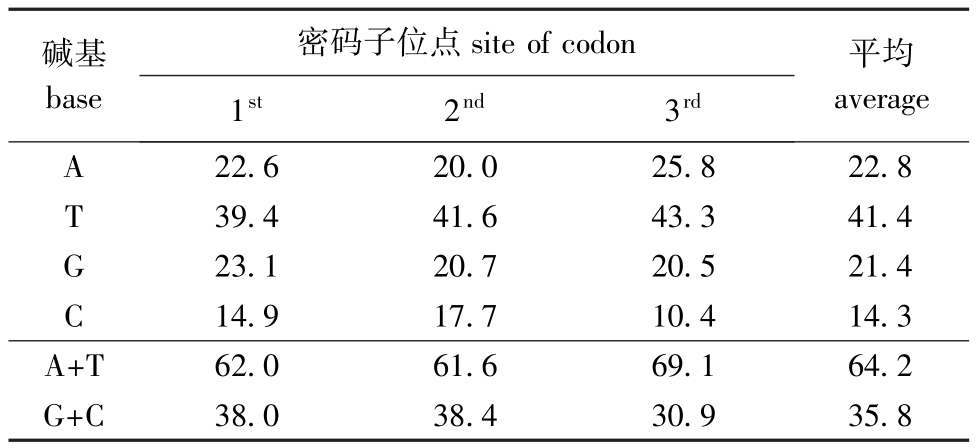
碱基base密码子位点site of codon 1st2nd3rd平均average A 22.6 20.0 25.8 22.8 T 39.4 41.6 43.3 41.4 G 23.1 20.7 20.5 21.4 C 14.9 17.7 10.4 14.3 A+T 62.0 61.6 69.1 64.2 G+C 38.0 38.4 30.9 35.8
本研究中的110条帘蛤目COI序列的核苷酸变异情况如表4所示。
2.2 种内和种间的遗传距离
基于K2P算法计算出110个个体的帘蛤目COI基因序列的种内和种间遗传距离如表5所示。16个物种的种内平均遗传距离为0.010 6,56.25%的种内遗传距离小于0.010 0,其中裂纹哥特蛤的种内遗传距离最大,为0.018 2,并未超出DNA条形码COI基因种类遗传距离2%的界限。
表4 16种帘蛤目贝类的COI部分序列密码子的变异
Tab.4 The variation in some codons of COI gene sequences of 16 species in Veneroida
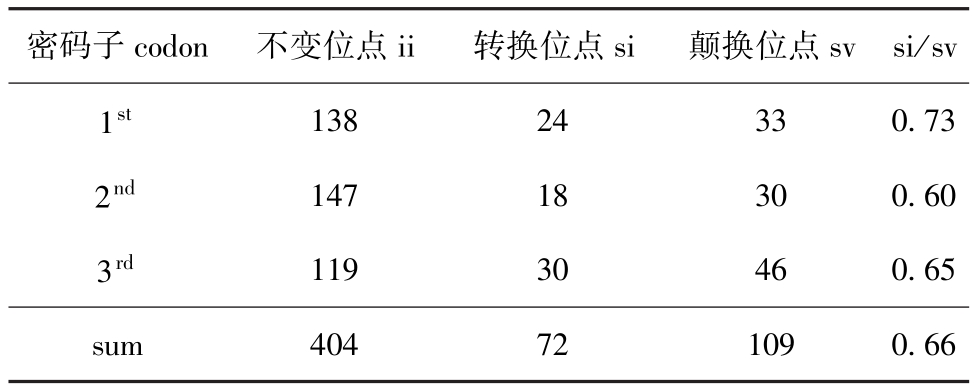
密码子codon 不变位点ii 转换位点si 颠换位点sv si/sv 1st138 24 33 0.73 2nd147 18 30 0.60 3rd119 30 46 0.65 sum 404 72 109 0.66
2.3 帘蛤目的分类及系统发育关系
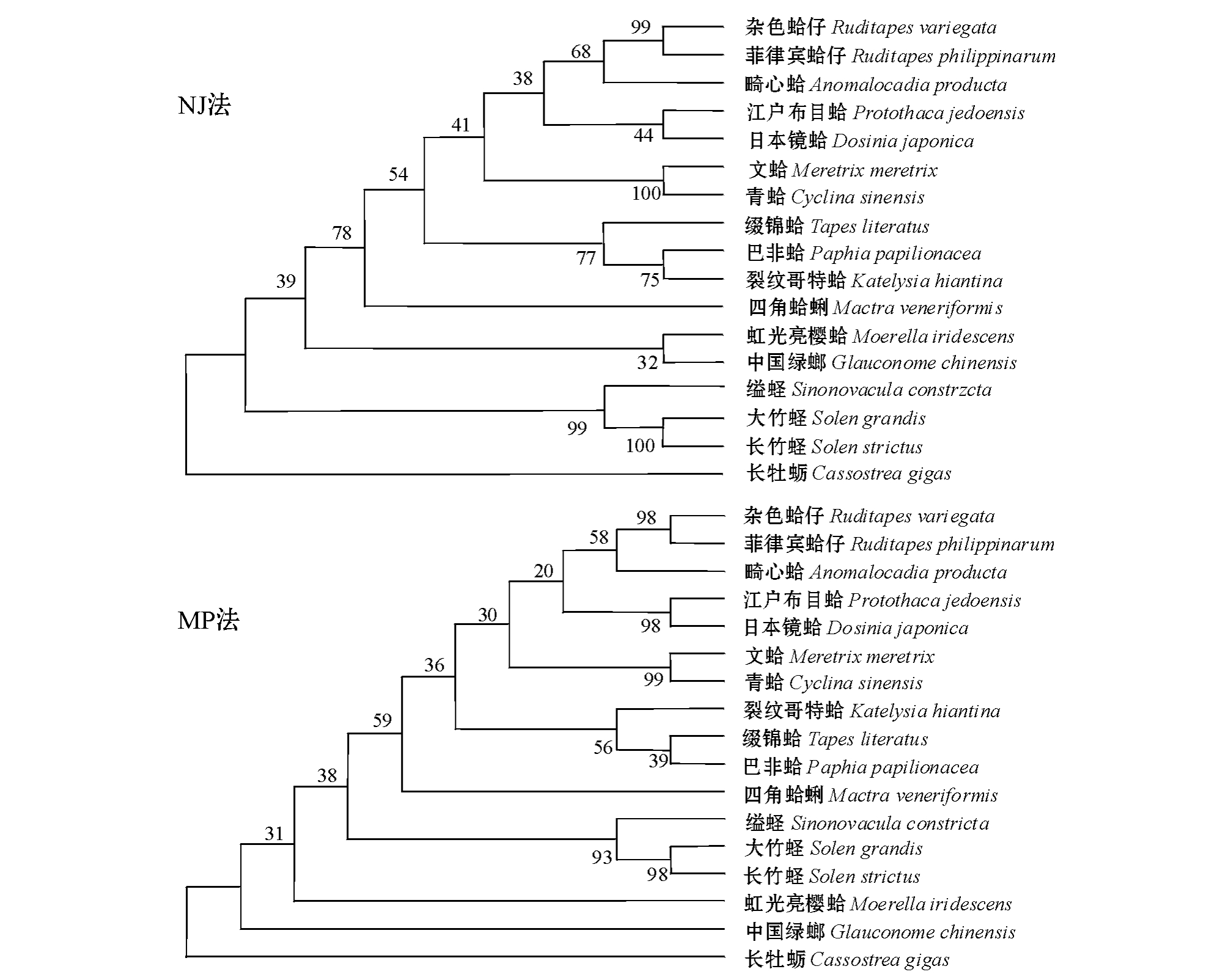
从基于NJ法和MP法构建的帘蛤目系统发育树(图1)可见,用两种方法构建的系统进化树得到相似的拓扑结构,但对不同分类阶元而言,分子系统关系与传统形态学分类结果不尽相同。在种水平上,NJ树和MP树的分类一致性均为100%;在属水平上,分子系统与形态学的分类结果略有差异。
表5 16种帘蛤目贝类的种间和种内遗传距离
Tab.5 Genetic distances in intraspecific and interspecies in 16 species in Veneroida

GP1 GP2 GP3 GP4 GP5 GP6 GP7 GP8 GP9 GP10 GP11 GP12 GP13 GP14 GP15 种内intraspecies GP1 0.0047 GP2 0.1905 0.0143 GP3 0.1447 0.1331 0.0059 GP4 0.2798 0.2693 0.1760 0.0172 GP5 0.2539 0.2956 0.2702 0.0646 0.0105 GP6 0.2220 0.2544 0.2530 0.2685 0.2159 0.0078 GP7 0.3443 0.3507 0.3047 0.3355 0.3229 0.3704 0.0047 GP8 0.3425 0.2977 0.3509 0.3443 0.3879 0.3297 0.3213 0.0122 GP9 0.2205 0.3653 0.3759 0.3239 0.3852 0.3810 0.3749 0.3642 0.0071 GP10 0.3524 0.3493 0.3898 0.3705 0.3152 0.2108 0.3419 0.3137 0.4019 0.0119 GP11 0.3356 0.3221 0.3286 0.3065 0.3880 0.3269 0.3281 0.2913 0.3449 0.3162 0.0065 GP12 0.3654 0.3591 0.3449 0.3705 0.3924 0.3209 0.3952 0.3118 0.3126 0.4646 0.4377 0.0023 GP13 0.2524 0.2379 0.4127 0.3384 0.4086 0.3197 0.3176 0.2136 0.2954 0.3042 0.3119 0.2102 0.0182 GP14 0.4219 0.3904 0.3473 0.4254 0.4186 0.5575 0.3695 0.3345 0.4668 0.4912 0.4958 0.4145 0.2998 0.0065 GP15 0.4383 0.3589 0.5832 0.4578 0.4430 0.5374 0.3831 0.3596 0.5520 0.5253 0.4771 0.4971 0.3138 0.3016 0.0055 GP16 0.5614 0.1899 0.5388 0.4509 0.4326 0.6401 0.6140 0.5535 0.5820 0.7270 0.5759 0.5493 0.4418 0.5820 0.5762 0.0108
3 讨论
3.1 帘蛤目线粒体COI基因的结构特征
本研究结果表明,在帘蛤目16个物种中有6个物种COI基因存在碱基插入和缺失现象,占到了37.50%。共存在103个插入和缺失位点,其中杂色蛤仔、裂纹哥特蛤插入和缺失位点均多达30个,大竹蛏插入和缺失位点为27个。碱基的插入和缺失现象在软体动物中较为常见[31-36],这可能与软体动物进化过程中线粒体拷贝数的变化有一定关联。帘蛤目贝类COI基因碱基组成中也存在着很普遍的偏倚现象。朱立静等[37]对四角蛤蜊COI基因的研究表明,四角蛤蜊的A+T含量明显高于G+C含量;程汉良等[38]对帘蛤目中6种贝类进行COI序列的扩增结果也表明,A+T含量明显高于G+C含量。这与本研究结果相似。线粒体碱基组成的偏倚现象符合无脊椎动物线粒体基因组的特点,其他无脊椎动物如虾、蟹、头足类等均存在这种相似的现象[39]。
COI基因存在着大量序列变异,适于用其进行群体遗传学多样性分析。在线粒体序列变异中,变异位点的转换较易在近缘种间频繁发生,而颠换在较远缘种间频繁发生;在同种动物中,转换往往在数量上远超过颠换。本研究中的物种分别属于不同种属关系,故颠换多于转换。
3.2 DNA条形码与形态学方法在帘蛤目贝类物种鉴定中的异同
DNA条形码对物种的鉴定可以弥补传统形态学鉴定存在的不足,为形态学鉴定提供充足的理论依据[26]。徐凤山[1]用形态学鉴定方法,将本研究中的16个种分成了6科,其中帘蛤科有菲律宾蛤仔、杂色蛤仔、畸心蛤、江户布目蛤、日本镜蛤、文蛤、青蛤、缀锦蛤、巴非蛤和裂纹哥特蛤;蛤蜊科有四角蛤蜊;樱蛤科有虹光亮樱蛤;绿螂科有中国绿螂;竹蛏科有大竹蛏和长竹蛏;截蛏科有缢蛏。本研究中构建的NJ系统树在科水平上与用形态学方法鉴定的一致性为83.3%,其中虹光亮樱蛤与中国绿螂分别属于不同的科,却聚为一支,而在MP树中则不存在这个问题;在属水平上,相同属的种类可以很好地聚为一支,一致性为100%。本研究中发现,青蛤与文蛤聚为一支,缀锦蛤、巴非蛤与裂纹哥特蛤聚为一支,大竹蛏、长竹蛏与缢蛏聚为一支,这可以说明他们之间的亲缘关系较近,这与形态学分类结果相符合[1],这样就为以往的形态学分类增加了可靠的依据。
3.3 DNA条形码在帘蛤目贝类物种鉴定中的适用性和局限性
本研究中所开发的全部条形码序列逐一与生物条形码数据库 (BoLD)中已经公布的生物条形码进行比对,匹配率高达100%,并与形态学鉴定相吻合,表明DNA条形码在帘蛤目贝类鉴定方面具有较高的适用性,完全可以通过COI基因序列的对比完成物种的准确鉴定。同时Hebert等[26]对动物界11个门13 320个物种的COI基因进行分析,发现其序列间的差异能够很好地区分所有研究物种,并认为在动物界中COI基因是合适的DNA条形码标准基因。Vences等[40]于 2005年探讨了DNA条形码对两栖爬行类物种的鉴定能力,结果显示选用的COI基因能够准确辨别各物种。此外Vences等[40]选用16S rRNA基因进行条形码分析,发现在两栖爬行类中16S rRNA基因具有较强的引物适应性,并且序列相对保守。对于国际上分类混乱的两栖爬行类,使用DNA条形码技术,不仅能够准确地鉴别出各物种,而且还能准确无误地识别出其不同地理类群的变异。对北美哥斯达黎加地区1 000多种鳞翅目昆虫生物多样性的调查中,Hajibabaei等[41]利用DNA条形码的COI基因序列建立了一个分类学平台,能够鉴定物种以及发现这一区域存在的模糊种,并且能准确鉴定97%的物种,区分不能被确认的物种是亲缘关系极近的物种或形态定义模糊的物种,由此认为,DNA条形码能够很好地协助进行一个地区的生物多样性调查。Hebert等[26]利用COI基因序列对北美260余种鸟类进行了有效区分。利用DNA条形码也能够对鱼类以及热带鳞翅目昆虫进行有效的物种鉴别。
Hebert等[26]指出,利用COI序列有效地进行物种鉴别的关键是种间的遗传距离必须大于种内的遗传距离。Hebert等[26]在对动物界11个门13 320个物种进行研究后得出,物种种内的遗传距离很少有大于2%的,大部分物种种内遗传距离小于1%。本研究结果表明,16种帘蛤目贝类种间的平均遗传距离为0.388 4,种内平均遗传距离为0.010 6,种间平均遗传距离为种内平均遗传距离的21.34倍;而种内遗传距离介于0.002 3~0.018 2,均小于2%,与Herbert等提出的观点相符。
COI适用于物种鉴定的原因在于COI基因既保证了足够多变异的同时又很容易被通用引物进行扩增,而且发生插入和缺失的也是很小的一部分序列,甚至不会发生插入和缺失现象。本研究中发现帘蛤目的贝类有一些碱基插入和缺失现象,孔晓瑜等[42]的研究发现,魁蚶也存在碱基的插入和缺失现象。而且这种插入和缺失的现象普遍存在于其他各种动物体中。
虽然基于COI基因的DNA条形码可以较好地实现物种的鉴定,但DNA条形码仍存在一定的缺陷,不可以完全取代形态学方法在分类学上的地位。DNA介导的物种鉴定重点在于种内和种间遗传差异的限定。种内和种间变异范围都不明确,而且不同物种的变异范围也不相同。就线粒体DNA而言,种内差异通常小于种间差异,但也有一些特例[34]。由于不同的种系间线粒体基因的水平转移,近缘物种共享线粒体多态性以及同属物种间的线粒体DNA修剪机制,都将造成快速分化物种与通过杂交产生的新物种在鉴定上的困难。本研究结果表明,DNA条形码在帘蛤目贝类的鉴定中有一定的适用性,但仍有许多问题需要解决。
参考文献:
[1] 徐凤山.中国海洋双壳类软体动物[M].北京:科学出版社, 1997:100-123.
[2] 齐钟彦.中国经济软体动物[M]北京:中国农业出版社,1996: 218-220.
[3] 齐钟彦.黄渤海的软体动物[M]北京:农业出版社,1987:146-150.
[4] Hebert PD N,Ratnasingham S,DeWard JR.Cytochrome c oxidase subunit Idivergences among closely related species[J].Barcoding Animal Life,2003,270(1):96-99.
[5] Ball S L,Hebert P D N,Burian S K.Biological identifications of mayflies Ephemeroptera using DNA barcodes[J].Journal of the North American Benthological Society,2005,24(3):508-524.
[6] Maynard B T,Kerr L J,Mckiernan JM,et al.Mitochondrial DNA sequence and gene organization in the Australian blacklip abaloneHaliotis rubra(Leach)[J].Mar Biotechnol,2005,7(1):645-658.
[7] Milbury C A,Gaffney PM.Completemitochondrial DNA sequence of the eastern oysterCrassostrea virginica[J].Mar Biotechnol, 2005,7(2):697-712.
[8] Passamonti M,Boore J L,Scali V.Molecular evolution and recombination in gender associated mitochondrial DNAs of the Manila clamTapes philippinarun[J].Genetics,2003,164(2):603-611.
[9] 陈丽梅,孔晓瑜,喻子牛,等.3种蛏类线粒体16S rRNA和COI基因片段的序列比较及其系统学初步研究[J].海洋科学, 2005,29(8):27-32.
[10] Park JK,Kim W.TwoCorbicula(Corbieulidae:Bivalvia)mitochondrial lineages arewidely distributed in Asian freshwater environment[J].Molecular Phylogenetics and Evolution,2003,29 (3):529-539.
[11] Pfenninger K,Reinhardt F,Streit B.Evidence for cryptic hybridization between different evolutionary lineages of the invasive clam genusCorbicula(Veneroida,Bivalvia)[J].Evol Biol, 2002,15(1):818-829.
[12] Smith P J,Mcveagh SM,Won Y,et al.Genetic heterogeneity among New Zealand speciesof hydrothermal ventmussels(Mytilidae:Bathymodiolus)[J].Marine Biology,2004,144(1):537-545.
[13] Therriault TW,Docker M F,Rlova M L,et al.Molecular resolution of the family Dreissenidae(Mollusca:Bivalvia)with emphasis on Ponto-Caspian species,including first report ofMytilopsis leucophaeatain the Black Sea basin[J].Molecular Phylogenetics and Evolution,2004,30(1):479-489.
[14] King TL,EackiesM S,Gjetvaj B,etal.Intraspecific phylogeography ofLasmigona subviridis(Bivalvia:Unionidae):conservation implications of range discontinuity[J].Molecular Ecology,1999, 8:S65-S78.
[15] Nagashima K,Sato M,Kawamata K,et al.Genetic structure of Japanese scallop population in Hokkaido analyzed by mitochondrial haplotype distribution[J].Mar Biotechnol,2005,7:1-10.
[16] Stepien C A,Morton B,Dabrowska K A,et al.Genetic diversity and evolutionary relationships of the troglodytic living fossilCongeri akusceri(Bivlvia:Dreissenidae)[J].Molecular Ecology, 2001,10(1):1873-1879.
[17] An H S,Jee Y J,Min K S,et al.Phylogenetic analysis of six species of Pacific abalone(Haliotidae)based on DNA sequences of 16S rRNA and cytochrome oxidase subunit Imitochondrial genes [J].Mar Biotechnol,2005,7(1):373-380.
[18] Carlini D B,Young R E,Vecchione M.A molecular phylogeny of the Octopoda(Mollusca:Cephalopoda)evaluated in light ofmorphological evidence[J].Molecular Phylogenetics and Evolution, 2001,21(3):388-397.
[19] Donald KM,KennedyM,Spencer H G.The phylogeny and taxonomy of Austral monodontine topshells(Mollusca:Gastropoda: Trochidae),inferred from DNA sequences[J].Molecular Phylogenetics and Evolution,2005,37(2):474-483.
[20] Goffredi SK,Hurtado L A,Hallam S,et al.Evolutionary relationships of deepsea ventand cold seep clams(Mollusca:Vesicomyidae)of the species complex[J].Marine Biology,2003,142(2): 311-320.
[21] Lin X,Zheng X,Xiao S,etal.Phylogeny of the cuttlefishes(Mollusca:Cephalopoda)based on mitochondrial COIand 16S rRNA gene sequence data[J].Acta Oceanologica Sinica,2004,23(4): 699-707.
[22] Machordom A,Araujo R,Erpenbeck D,et al.Phylogeography and conservation genetics of endangered European Margaritiferidae (Bivalvia:Unionoidea)[J].Biological J of the Linnean Society, 2003,78(2):235-252.
[23] Minton R L,Lydeard C.Phylogeny,taxonomy,geneticsand global heritage ranks of an imperiled freshwater snail genusLithasia(Pleuroceridae)[J].Molecular Ecology,2003,12(2):75-87.
[24] Nikula R,V In L R.Phylogeography ofCerastodermaglaucum(Bivlvia:Cardiidae)across Europe:amajor break in the EasternMediterranean[J].Marine Biology,2003,143(2):339-350.
[25] Folmer O,Black M,Hoeh W,et al.DNA primers for amplification ofmitochondrial cytochrome c oxidase subunit I from divercemetazoan invertebrates[J].Mol Mar Biol Biotech,1994,3(3):294-299.
[26] Hebert PD,Cywinska A,Ball SL,et al.Biological identifications through DNA barcodes[J].Proc Biol Sci,2003,270:313-321.
[27] Kumar S,Tamura K,NeiM.MEGA 3:Integrated software formolecular evolutionary genetics analysis and sequence alignment [J].Briefings in Bioinformatics,2004,5(2):150-163.
[28] Rozas J,Sánchez-DelBarrio JC,Messeguer X.DnaSPDNA polymorphism analyses by the coalescent and other methods[J]. Bioinformatics,2003,19(2):2496-2497.
[29] 徐钢春,魏广莲,李建林,等.基于线粒体DNA D-loop序列分析养殖刀鲚与湖鲚的遗传多样性[J].大连海洋大学学报, 2012,27(5):448-452.
[30] Boore JL.Complete DNA sequence of themitochondrial genome of the black chiton,Katharina tunicate[J].Genetics,1994,138: 423-443.
[31] Boore J L.Animal mitochondrial genomes[J].Nucleic Acids Res,1999,27(8):1767-1780.
[32] Lindgren R A.Molecular inference of phylogenetic relationships among Decapodiformes(Mollusca:Cephalopoda)with special focus on the squid order Oegopsida[J].Mol Phylogen Evol,2010, 56(1):77-90.
[33] Akasaki T,Nikaido M,Tsuchiya K,et al.Extensivemitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications[J].Mol Phylogen Evol,2006,38(3):648-658.
[34] Brown W M,George JM,Wilson A C.Rapid evolution of animal mitochondrial DNA[J].Proc Natl Acad Sci USA,1979,76(3): 1967-1971.
[35] Masaaki Y,Kazuhiko T,Hisetaka F.Phylogeny of selected Sepiidae(Mollusca,Cephalopoda)based on 12S,16S,and COI sequence,with comments on the taxonomy reliability of severalmorphological characters[J].Zoolog Sci,2006,23:341-351.
[36] Zheng X D,Xiao S,Chen B,et al.Study on COI and 16S rRNA gene sequences of the common Chinese cuttlefishSepiella maindroni[J].Transaction of the Chinese Society of Malacology, 2001,9:64-69.
[37] 朱立静,陈淑吟,许晓风,等.四角蛤蜊江苏群体线粒体COI基因片段序列研究[J].江苏农业科学,2010(4):33-35.
[38] 程汉良,吴婷婷,夏德全,等.6种帘蛤科贝类及4个地理种群文蛤线粒体COI基因片段序列分析[J].海洋学报,2007 (5):109-116.
[39] Yancheng.Relationships of the genusMeretrix(Mollusca:Veneridae)based on mitochondrial COI gene sequences[J].Zoology Res,2009,30(3):233-239.
[40] Vences M,Thomas M,Bonett R M,et al.Deciphering amphibian diversity through DNA barcoding:chances and challenges[J]. Phil Trans R Soc B,2005,360:1859-1868.
[41] HajibabaeiM,Janzen D H,Burns JM,et al.DNA barcodes distinguish species of tropical lepidoptera[J].Proc Natl Acad Sci USA,2006,103(4):968-971.
[42] 孔晓瑜,姜艳艳,相建海,等.魁蚶线粒体16S rRNA和COI基因片段序列测定及其应用前景[J].海洋科学,2001,25(12): 46-48.
DNA barcoding and molecular phylogeny of 16 species of economical shellfish in Veneroida along China Coast
WANG Lin-nan1,YAN Xi-wu1,QIN Yan-jie1,NIE Hong-tao1,NIU Hong-bo1,ZHANG Guo-fan2
(1.Engineering Research Center of Shellfish Culture and Breeding in Liaoning Province,College of Fisheries and Life Science,Dalian Ocean University,Dalian 116023,China;2.Research and Development Center of Marine Biotechnology,Institute of Oceanology,Chinese Academy of Sciences,Qingdao 266071,China)
Abstract:Mitochondrial COI genes were sequenced from 60 individuals of 6 common species in Veneroida along China coast using the universal barcoding primers,and the 60 sequences were compared with the other 50 homologous sequences from 10 shellfish species in Veneroida derived from the GenBank.The results showed that there were Insert-Deletion(indel)sites in many COI sequences in Veneroida species,total indel siteswere 103,including COI sequence indels of up to 30 for Manila clamRuditapes variegateandKatelysia hiantina,and 27 forSolengrandis.On average,the content of A+T(64.2%)was found significantly higher than that of G+C(35.8%). The Kimera-2-parameter model indicated that there was mean distances of 0.388 4 in pairwise-species and 0.010 6 in species.According tomaximum parsimony and neighbor joining trees for all 110 sequences of Veneroida,there was very high consistency between molecular analysis and morphological classification on family,genus, and species levels.Consequently,the findings provided essential revision and supplement to morphology taxonomy.
Key words:Veneroida;COI gene;phylogeny;taxonomy;DNA barcoding
中图分类号:S917.4
文献标志码:A
文章编号:2095-1388(2013)05-0431-07
收稿日期:2013-02-21
基金项目:现代农业产业技术体系建设专项资金资助项目 (CARS-48)
作者简介:王琳楠 (1988-),女,硕士研究生。E-mail:xlypt@126.com
通信作者:闫喜武 (1962-),男,博士,教授。E-mail:Yanxiwu2002t@163.com