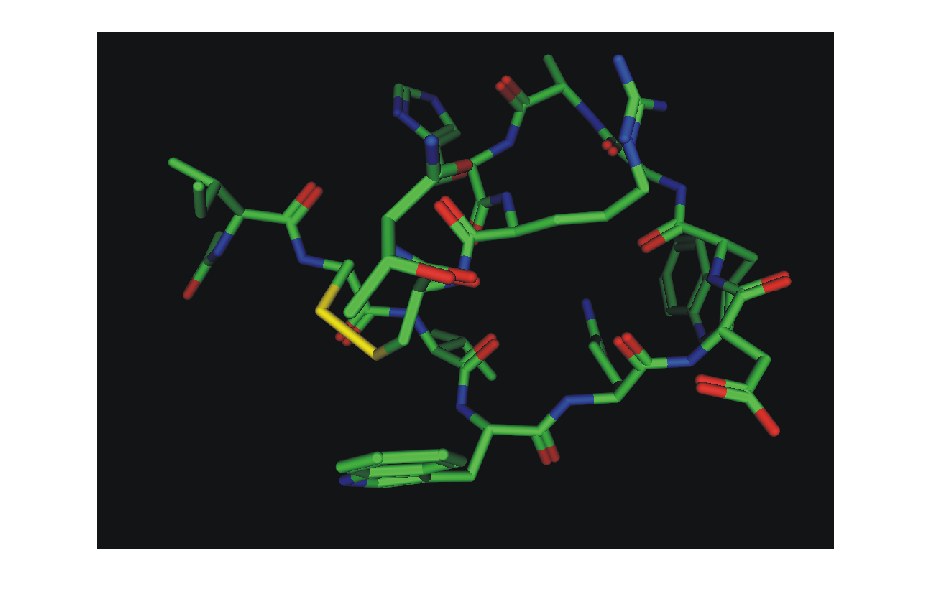
图1 抑制剂分子Compstatin的初始结构
Fig.1 The initial conformation of Compstatin
摘要:采用分子动力学模拟取样,运用MM-PBSA方法计算了人类C3c-Compstatin复合物的结合自由能。通过能量分解方法探究了人类补体蛋白C3c上抑制剂结合域MG4~MG5(巨球蛋白区域)上的主要残基与配体Compstatin纤维蛋白之间的相互作用和识别。结果表明:C3c与补体抑制剂Compstatin的理论结合自由能(-8.06 kcal/mol)与试验值(-6.72 kcal/mol)吻合较好,分子内能总和(-177.24 kcal/mol)对补体抵制剂的结合贡献最大,其次为真空静电作用能(-108.74 kcal/mol)和范德华作用能(-68.51 kcal/mol)。作为对结晶试验的补充,进行了C3c蛋白和小分子抑制剂之间结合的动态过程以及在两者结合的影响下蛋白形态变化的分子动力学模拟,结果发现C3c上的残基ARG459、ASP491、MET457和Compstatin上的残基TRP7、TRP4、HIS10为结合自由能作出了主要贡献。该结果对进一步研究C3补体抑制剂的机制提供了结构方面的信息。
关键词:分子动力学;MM-PBSA/GBSA方法;结合自由能;补体蛋白C3c;结合肽
在人体免疫通路中,补体蛋白C3在细菌或炎灶分解产物的作用下裂解出C3c,C3c是一种参与免疫激活通路中必不可少的蛋白。但是C3c多度表达会引发诸多复杂疾病或症状,如系统性红斑狼疮、重症肌无力、肺出血和肾炎综合征等疾病,以及异种移植超急性排斥反应等症状。目前,几种C3补体的抑制剂仍处于研发状态,尚未在临床试验中应用[1-10]。研发内容以CR1(补体受体)补体活化调节剂以及对其进行修饰的研究居多,但其结果对一些疾病的疗效却不尽如人意。近年来发现的小分子补体抑制剂与上述传统生物大分子相比,在药物方式、药物动力学方面均有许多优点。Morikis等[11]认为,C3结合肽(Compstatin)可能会通过抑制剂C3转化酶(C3bBb)的形成和降低C3转化酶的稳定性来抑制C3的活化,但最新研究[12-13]推测,它是通过结合到C3分子的C3c部分来阻止C3裂解成C3a和C3b,从而抑制C3的活化。Compstatin是一种带13个氨基酸(ICVVQDWGHHRCT-NH2,在Cys-2-Cys-12有二硫键)的环状纤维蛋白多肽(图1),它可以有效抑制C3裂解反应中C3转变成C3b,使之转变成C3c-Compstatin复合物,以阻止C3b进一步与自身物质——抗体结合而提供强烈的清除自身靶点复合物的信号。Compstatin因具有抑制效果明显、分子量小和非免疫原性等特点[14],已经成为一种新的补体抑制剂潜药。目前,Compstatin与C3蛋白结合的机制尚不明确,因此,进一步研究Compstatin与C3蛋白的结合十分必要。本研究中将针对C3c蛋白与抑制剂的结合进行模拟研究。
图1 抑制剂分子Compstatin的初始结构
Fig.1 The initial conformation of Compstatin
1.1 C3c-Compstatin的结构与模型的选取
由于酶的变构抑制效应,Compstatin在水溶液中的自由状态与在结合于C3上的状态不同。本研究中,复合物初始结构为C3c-Compstatin的结晶结构(PDB ID 2qki)取自于蛋白质库(http:// www.rcsb.org/pdb/),分辨率为2.4 Å,C3c是C3主要的多肽片段,它由β链(23~665个氨基酸)巨球蛋白域MG1~MG7、α链(749~936)锚定区域 anchor、α链(1321~1663)C345c区域组成,其中与抑制剂Compstatin结合的是β链上的MG巨球蛋白域中的MG4~MG5区域,因为侧链anchor和C345c区域远离配体结合位点,为了节省计算时间和资源,模型不包括α链(749~936)锚定区域anchor和α链(1321~1663)C345c区域。利用分子动力学方法补齐结晶体缺失的残基后,制成分子动力学的输入文件。
1.2 分子动力学模拟
分子动力学模拟采用 Amber 11程序包中的Sander及Pmemd模块[15],受体和配体蛋白全部采用ff99[16]力场。若把整个体系放入水中,总原子数将超过Amber 11程序能够允许的范围,并且在模拟准确性下降的同时还会严重影响计算机模拟的时间,为此,采用在距抑制剂Compstatin质心2.0 nm的复合物上包一个 “水帽”(solvate cap)的方式加水。水溶液采用显性的TIP3P模型,为了使整个系统总体电荷为0,添加了1个钠离子,分子动力学初始结构如图2所示。用SHAKE法来限制所有含氢键的伸缩[17],模拟步长取2 fs。非键相互作用的截断值(Cutoff)为1.2 nm,在运行分子动力学之前,根据 Ryckaert等[17]的研究结果,先用5 000步最陡下降法(steepest descent method),紧接着用5 000步共轭梯度法(conjugate gradient)来消除分子间的高能碰撞;然后约束溶质部分,设定所有水分子及抗衡离子可以移动,将体系升温到300 K,使其在密度为1 g/cm3时保持平衡;取消溶质约束,利用NTP系宗进行分子动力学模拟,产生10 ns的生产相轨迹,只对后1 ns平衡构象进行采样,每2 ps取一次构象,利用采集的构象进行自由能计算。
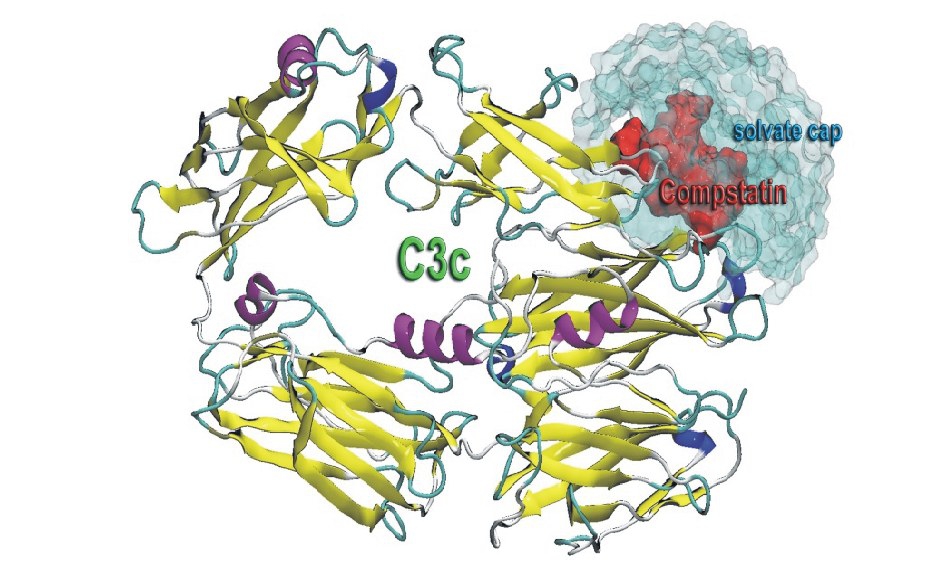
图2 分子动力学初始结构
Fig.2 The initial conformation in molecular dynamics
注:C3c蛋白由卡通结构显示,Compstatin由红色表面显示,水帽子则由青色半透明表面显示。
Note:Cartoon structure of C3c,Compstatin is shown in red surface, and water cap is shown in cyan trans-lucent surface.
1.3 自由能的计算公式
采用Amber 11程序包中的 mm_pbsa.py模块,使用 MM/PBSA 方法计算结合自由能△G[18-21],其计算公式为
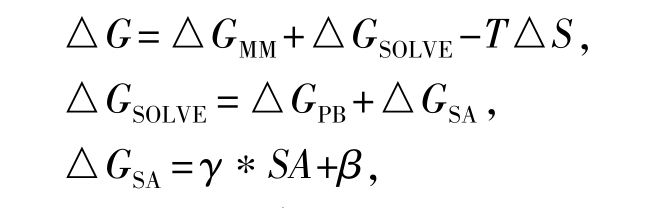
其中:△GMM为分子真空下的内能差,通过分子力学方法获得,包括真空静电相互作用能△GELE和真空范德华作用能△GVDW;△S为通过正则模拟方法计算获得的熵变;T为绝对温度;△GSOLVE为溶剂化自由能差;△GPB为通过求解Poisson-Boltzmann方程[22]获得的极性溶剂化自由能差;GSA为非极性溶剂化自由能差;SA为溶剂可及表面的表面积,采用Tsui和 Case等建立的参数,γ和 β分别为0.005 42 kcal/(mol·Å)和0.92 kcal/mol[23],在计算中,溶剂采用介电常数为78的连续介质模型,溶质的介电常数视为1,原子的半径取PARSE参数序列。
PBSA计算中截取的是轨迹稳定状态的200帧构象,由于利用正则模拟的Nmode模块计算熵变非常占内存[24],因此,取稳定状态的构象50帧,并且切除了距离补体抑制剂2.5 nm外C、N两末端不与配体发生作用的残基,在整个体系没有断裂共价键的构象下计算熵变[21]。
1.4 能量分解
本研究中能量分解采用 GBSA(Generalized Born/Surface Area)方法[25]来计算。C3c和Compstatin各个残基相互作用能的计算公式为
其中:△GVDW和△GELE分别为每个残基在真空中的范德华作用能和静电相互作用能;△GGB为每个残基的极性溶剂化能,该项由广义的波恩模型计算得到;△GGBsur为非极性溶剂化能,由LCPO模型计算得到。将每项能量均分解为残基的主链能量贡献和侧链能量贡献。
2.1 动力学轨迹
图3为C3c蛋白与抑制剂复合物的骨架原子(CA、N、C)在整个模拟过程中相对于初始结构随时间变化的均方根偏差(RMSD),可以看出在整个10 ns模拟过程中,RMSD涨落较小,波动范围为0~2 Å,在前3.5 ns中,曲线一直在波动中上升,之后进入稳定状态,RMSD于1.6 Å上下浮动,直到模拟结束时RMSD不超过2 Å,说明整个模拟体系是稳定的。因此,在最后0.4 ns的轨迹中选取200个构象用于自由能计算是合理的。
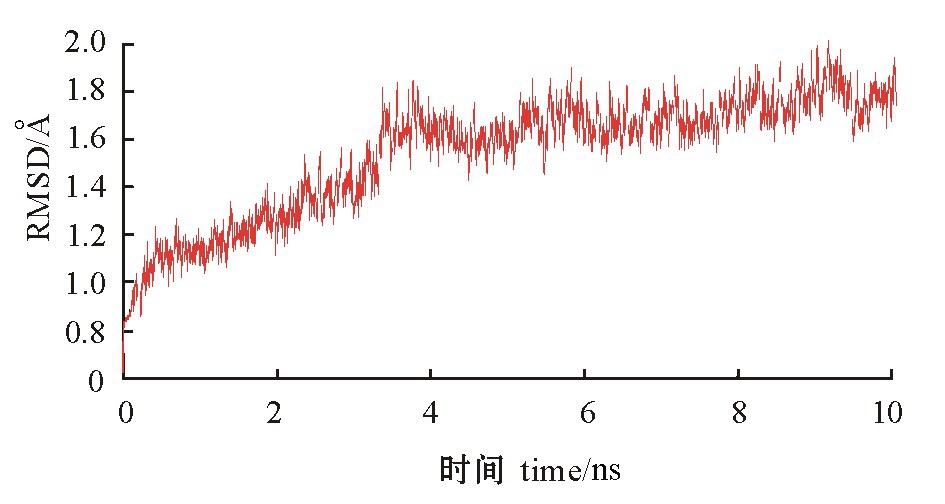
图3 C3c蛋白与抑制剂复合物的骨架原子(CA、N、C)的RMSD随时间的变化
Fig.3 Changes in RMSD of the back bone(CA,N, C)in the C3c-compstatin complex with time
2.2 自由能
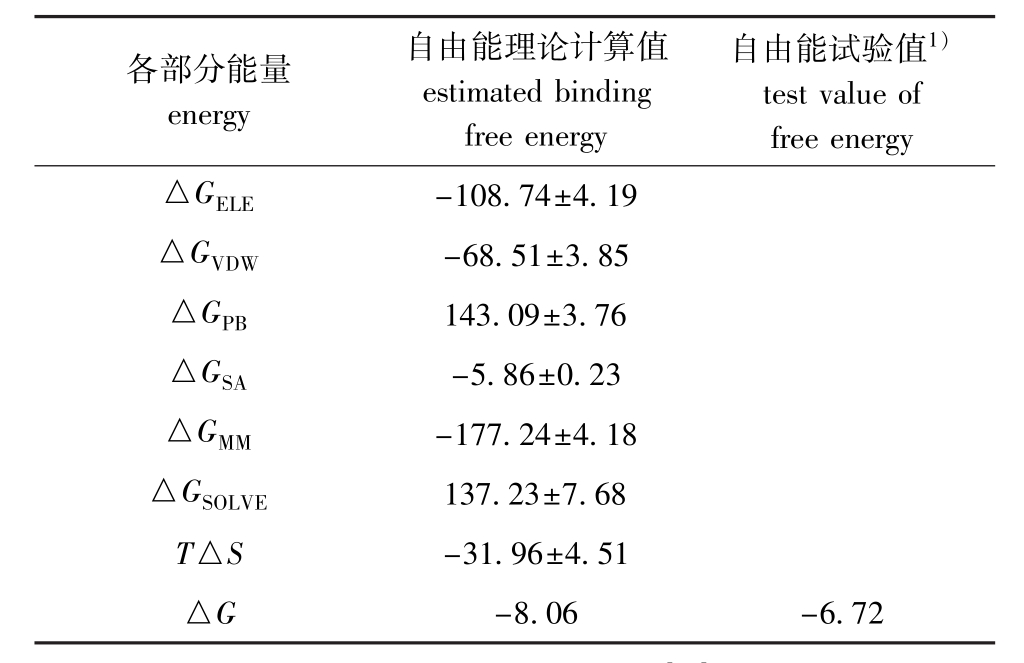
表1中列出了各项能量对吉布斯自由能的贡献。根据自由能的计算公式,可以得出 C3c与Compstatin的理论结合自由能为-8.06 kcal/mol,与试验值-6.72 kcal/mol[26]比较吻合,在形成复合物时,分子内能总和对配体结合的贡献最大,为-177.24 kcal/mol,真空静电作用能(-108.74 kcal/mol)和范德华作用能(-68.51 kcal/mol)也都有利于补体抑制剂的结合。
表1 C3c与Compstatin结合自由能的理论计算值和试验
值(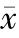 ±s)
±s)
Tab.1 Binding free energy estimated value and experi mental energy value between Compstatin and C3c kcal/mol
注:1)为由Ki值转换得到的结合能试验值[26]。
Note:1)The experimental data of binding energy converted from Ki.
各部分能量energy自由能理论计算值estimated binding free energy自由能试验值1)test value of free energy△GELE-108.74±4.19△GVDW-68.51±3.85△GPB143.09±3.76△GSA-5.86±0.23△GMM-177.24±4.18△GSOLVE137.23±7.68 T△S -31.96±4.51△G -8.06 -6.72
与众多文献一致[21,27-30],本研究中静电作用能总和(△GELE+△GPB)为34.35 kcal/mol,不利于配体的结合,主要表现在极性溶剂化能△GPB(143.09 kcal/mol)阻碍了复合物的形成,尽管真空静电作用能△GELE可以补偿大部分极性溶剂化能对补体抑制剂Compstatin结合所产生的不利影响,但对配体结合的阻碍仍然存在。
2.3 Compstatin与C3c的结合机制
由复合物的晶体结构可知,Compstatin与C3c形成了9对氢键[26]。以其作为对照比较,将模拟10 ns的生产相中受体与配体产生的氢键列于表2。从表2可见,大部分氢键理论计算值和结晶试验值相近,模拟过程中没有发现ACE0:O-ASN390:OD1和GLN5:NE2-ASP491:OD之间形成氢键,与结晶试验略有不同。模拟过程中显示存在两对存留时间很长的氢键,如图4-A中,一对为ALA9主链上的N与ASP491侧链羧基上的OD1形成了存活率为95.14%的氢键,说明这对氢键对抑制剂与C3c蛋白结合非常重要;另外一对为MET457:O与TRP7-NE1,MET457主链上的O充当氢受体,配体TRP7侧链上的NE1充当氢供体,这对氢键在模拟过程中存留时间约9 ns,也对稳定复合物的形成作出了有利贡献。
表2 10 ns模拟中C3c与抑制剂之间的氢键存活率和距离
Tab.2 Occupancy and distance of hydrogen bonds between C3c and Compstatin during 10 ns of MD simulation
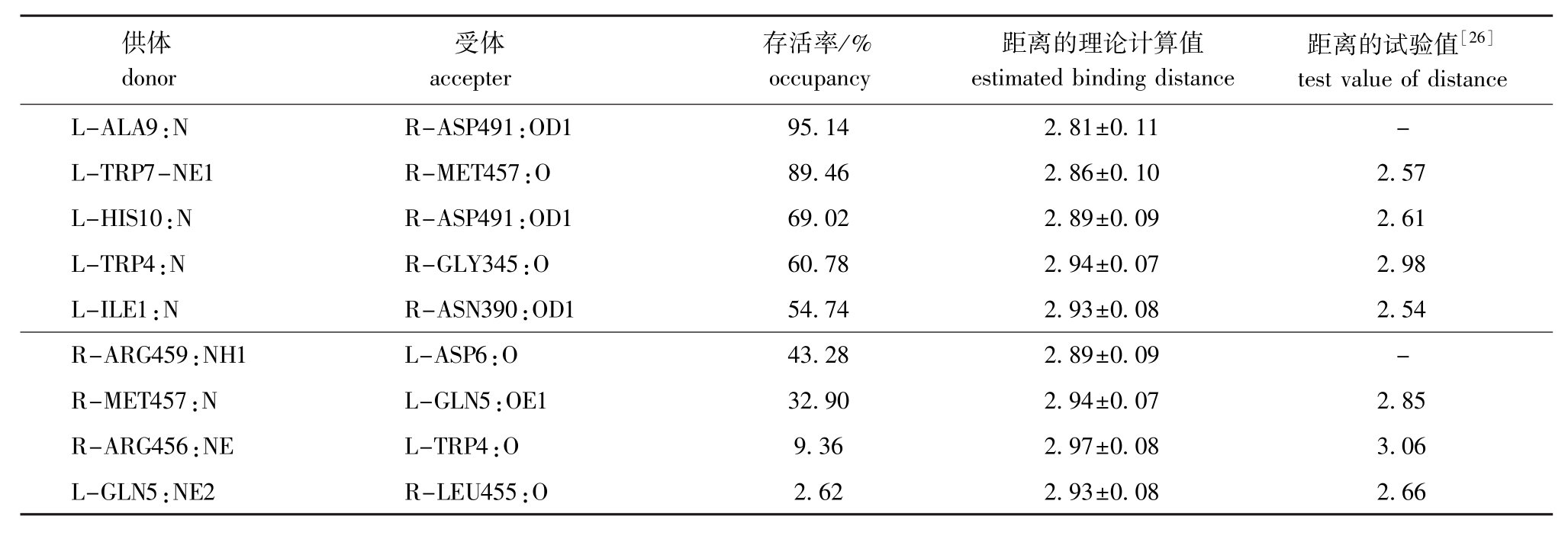
注:L表示配体抑制剂上的氨基酸残基;R表示受体C3c蛋白上的氨基酸残基。
Note:L represents the amino acid residues of the ligand inhibitor;R represents the amino acid residues of receptor C3c.
供体donor受体accepter存活率/% occupancy距离的理论计算值estimated binding distance距离的试验值[26]test value of distance L-ALA9:N R-ASP491:OD1 95.14 2.81±0.11 -L-TRP7-NE1 R-MET457:O 89.46 2.86±0.10 2.57 L-HIS10:N R-ASP491:OD1 69.02 2.89±0.09 2.61 L-TRP4:N R-GLY345:O 60.78 2.94±0.07 2.98 L-ILE1:N R-ASN390:OD1 54.74 2.93±0.08 2.54 R-ARG459:NH1 L-ASP6:O 43.28 2.89±0.09 -R-MET457:N L-GLN5:OE1 32.90 2.94±0.07 2.85 R-ARG456:NE L-TRP4:O 9.36 2.97±0.08 3.06 L-GLN5:NE2 R-LEU455:O 2.62 2.93±0.08 2.66
为了研究C3c蛋白与Compstatin结合的作用机制,本研究中使用Amber 11软件中的GBSA模块计算了抑制剂Compstatin与C3c上各残基的相互作用,其主要作用残基的能量分解见表3。从表3可见,C3c上的残基ARG459、ASP491和MET457与抑制剂Compstatin上的残基 TRP7、TRP4、HIS10对结合自由能的贡献较大。其中受体上每个残基对结合能的贡献比配体上的残基贡献要相对小一些。在受体上,虽然 ASP491具有最大的静电能(-22.19 kcal/mol),但由于侧链上较大的极性溶剂化能(23.17 kcal/mol)与其抵消,使 ASP491的贡献(-2.79 kcal/mol)在受体残基贡献中排列第二。受体上结合自由能贡献最大的是ARG459(-5.14 kcal/mol),同样发现其侧链上有巨大的极性溶剂化能(15.36kcal/mol) 与其静电能(-16.81 kcal/mol)抵消。而在配体中,残基TRP7的贡献力最大(-10.741 kcal/mol),主要来自其侧链上的范德华作用能(-8.80 kcal/mol)。正如在平均结构中发现的,整个TRP7的侧链像一把钥匙 一 样 陷 在 由 LEU492、 LEU454、 ASP491、GLY489、ARG456、MET457、ARG459组成的空腔中(图4-B),另外,配体中TRP7和TRP4的重要贡献与最近的试验结果吻合[31]。能量分解很好地验证了C3c和抑制剂Compstatin的结合模式。这些结果为基于C3c蛋白结构进行合理设计补体抑制剂提供了线索。
表3 用GB方法进行能量分解的主要贡献残基各部分的能量值
Tab.3 Energy contribution of the key residues computed by GB model kcal/mol
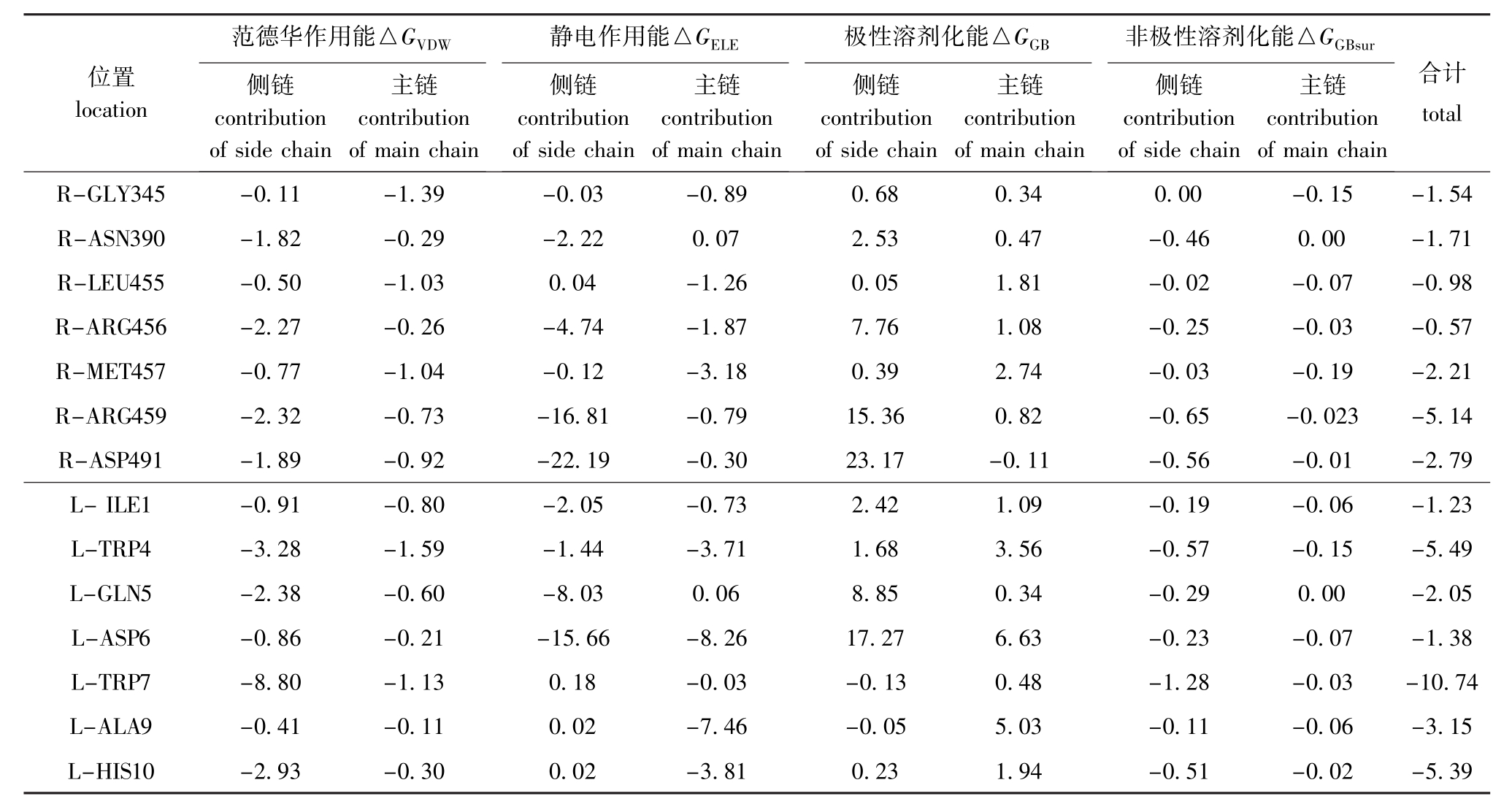
位置location范德华作用能△GVDW侧链contribution of side chain主链contribution of main chain静电作用能△GELE侧链contribution of side chain主链contribution of main chain极性溶剂化能△GGB侧链contribution of side chain主链contribution of main chain非极性溶剂化能△GGBsur侧链contribution of side chain主链contribution of main chain合计total R-GLY345 -0.11 -1.39 -0.03 -0.89 0.68 0.34 0.00 -0.15 -1.54 R-ASN390 -1.82 -0.29 -2.22 0.07 2.53 0.47 -0.46 0.00 -1.71 R-LEU455 -0.50 -1.03 0.04 -1.26 0.05 1.81 -0.02 -0.07 -0.98 R-ARG456 -2.27 -0.26 -4.74 -1.87 7.76 1.08 -0.25 -0.03 -0.57 R-MET457 -0.77 -1.04 -0.12 -3.18 0.39 2.74 -0.03 -0.19 -2.21 R-ARG459 -2.32 -0.73 -16.81 -0.79 15.36 0.82 -0.65 -0.023 -5.14 R-ASP491 -1.89 -0.92 -22.19 -0.30 23.17 -0.11 -0.56 -0.01 -2.79 L-ILE1 -0.91 -0.80 -2.05 -0.73 2.42 1.09 -0.19-0.06 -1.23 L-TRP4 -3.28 -1.59 -1.44 -3.71 1.68 3.56 -0.57 -0.15 -5.49 L-GLN5 -2.38 -0.60 -8.03 0.06 8.85 0.34 -0.29 0.00 -2.05 L-ASP6 -0.86 -0.21 -15.66 -8.26 17.27 6.63 -0.23 -0.07 -1.38 L-TRP7 -8.80 -1.13 0.18 -0.03 -0.13 0.48 -1.28 -0.03 -10.74 L-ALA9 -0.41 -0.11 0.02 -7.46 -0.05 5.03 -0.11 -0.06 -3.15 L-HIS10 -2.93 -0.30 0.02 -3.81 0.23 1.94 -0.51-0.02 -5.39
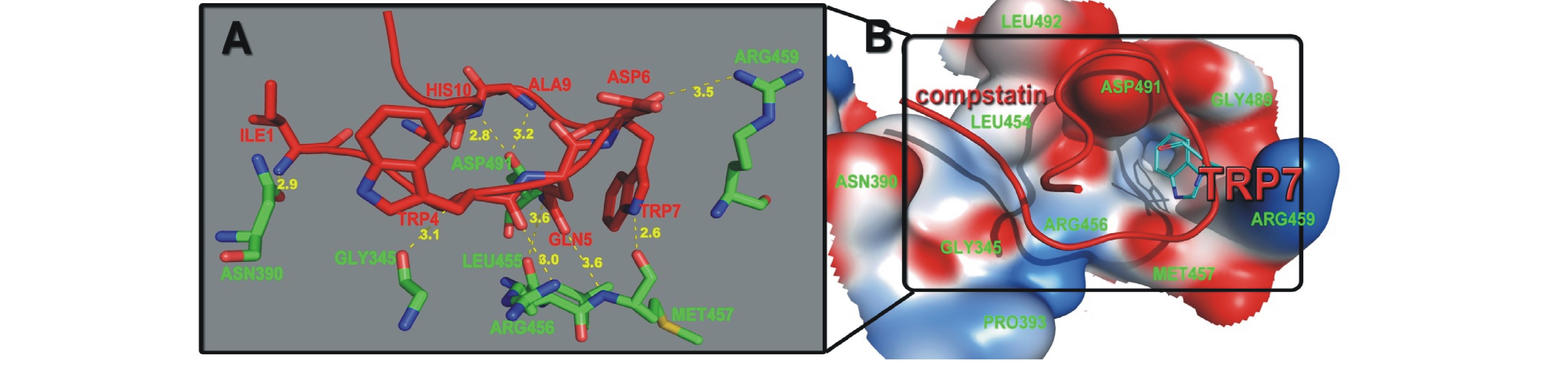
图4 C3c蛋白与抑制剂结合位点
Fig.4 The binding site of C3c-compstatin
注:A结合区域放大后的受体和配体中重要氨基酸残基以stick结构显示,红色碳骨架的是配体上的残基,绿色碳骨架的是受体上的残基;B红色ribbon显示的是Compstatin,受体结合域内的氨基酸以静电surface形式表现。
Note:A,Zooming in the binding region,all are shown as stick,carbon atoms on ligand are shown in red,and carbon atoms on receptor are shown in green;B,The red ribbon is Compstatin,and the global binding region on the receptor is shown by static surface.
本研究中采用了MM-PBSA结合自由能计算和GBSA能量分解的方法,经过10ns的分子动力学取样,计算了C3c蛋白和Compstatin复合物的结合作用能,计算的结合能结果与试验值吻合度较好,并且推断出对结合起关键作用的氨基酸残基。本研究表明,范德华作用能和非极性溶剂化能对复合物结合起主导作用,如真空静电作用能(-108.74 kcal/mol)和范德华作用能(-68.51 kcal/mol)为补体抑制剂结合提供主要贡献。研究中还发现:受体上ASP491参与形成的氢键时间最长(9.5 ns),能量分解中其静电能贡献也最大(-22.19 kcal/mol);配体上TRP7参与形成的氢键时间最长(8.9 ns),能量分解中其结合能贡献也最大(-10.74 kcal/mol)。从能量分解中可以看出,TRP7侧链的范德华作用力为-8.80 kcal/mol,再次说明范德华能对结合非常有利,而从平均结构中发现的TRP吲哚基团恰好插入在结合位点所形成的空腔中,也为能量计算进行了辅证。通过氢键分析和能量分解进一步推断出影响受体和配体结合的重要氨基酸 TRP7、TRP4及氢键 ASP491:OD1-ALA9:N和MET457:O-TRP7-NE1,这一结果填补了X-ray结晶试验所得不到的分子动态结合的信息,为设计高效的人类补体C3c蛋白抑制剂提供了重要的结构信息。
致谢:感谢大连理工大学李艳教授提供Amber软件!
参考文献:
[1]Thomas L J,Panneerselvam K,Beattie D T,et al.Production of a complement inhibitor possessing sialyl Lewis X moieties by in vitro glycosylation technology[J].Glycobiology,2004,14:883-893.
[2]Souza D G,Esser D,Bradford R,et al.APT070(Mirococept),a membrane-localised complement inhibitor,inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury [J].British Journal of Pharmacology,2005,145:1027-1034.
[3]Atkinson C,Song H,Lu B,et al.Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection[J].Journal of Clinical Investigation,2005,115(9):2444-2453.
[4]罗雪,汪正清.人sCR1结合C3b结构域基因的克隆及在大肠杆菌中高效表达[J].第三军医大学学报,2005,27:1551-1554.
[5]谭兵,兰阳军,张德纯,等.重组人可溶性补体受体 1型SCR15—18片段对心肌缺血再灌注损伤的保护作用[J].中华心血管病杂志,2008,35:1037-1040.
[6]Lazar H L,Bokesch P M,Van Lenta F,et al.Soluble human complement receptor 1 limits ischemic damage in cardiac surgery patients at high risk requiring cardiopulmonary bypass[J].Circulation,2004,110:II274-II279.
[7]Zimmerman J L,Dellinger R P,Straube R C,et al.Phase I trial of the recombinant soluble complement receptor 1 in acute lung injury and acute respiratory distress syndrome[J].Critical Care Medicine,2000,28:3149-3154.
[8]Sahu A,Lambris J D.Complement inhibitors:a resurgent concept in anti-inflammatory therapeutics[J].Immunopharmacology, 2000,49:133-148.
[9]Bureeva S,Andia-Pravdivy J,Kaplun A.Drug design using the example of the complement system inhibitors'development[J].Drug Discovery Today,2005,10:1535-1542.
[10]Holland M C H,Morikis D,Lambris J D.Synthetic small-molecule complement inhibitors[J].Current Opinion in Investigational Drugs,2004,5:1164-1173.
[11]Morikis D,Lambris J.Structural aspects and design of low-molecular-mass complement inhibitors[J].Biochemical Society Transactions,2002,30:1026-1036.
[12]Rooijakkers S H M,Wu J,Ruyken M,et al.Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor[J].Nature Immunology,2009,10:721-727.
[13]Katschke Jr K J,Stawicki S,Yin J,et al.Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement[J].Journal of Biological Chemistry,2009,284:10473-10479.
[14]Klepeis J L,Floudas C A,Morikis D,et al.Integrated computational and experimental approach for lead optimization and designof compstatin variants with improved activity[J].Journal of the A-merican Chemical Society,2003,125:8422-8423.
[15]Case D,Darden T,Cheatham T,et al.AMBER 11[M].San Francisco:University of California,2010.
[16]Wang J,Cieplak P,Kollman P A.How well does a restrained electrostatic potential(RESP)model perform in calculating conformational energies of organic and biological molecules?[J].Journal of Computational Chemistry,2000,21:1049-1074.
[17]Ryckaert J P,Ciccotti G,Berendsen H J C.Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of nalkanes[J].Journal of Computational Physics,1977,23:327-341.
[18]Kollman P A,Massova I,Reyes C,et al.Calculating structures and free energies of complex molecules:combining molecular mechanics and continuum models[J].Accounts of Chemical Research,2000,33:889-897.
[19]Srinivasan J,Cheatham III T E,Cieplak P,et al.Continuum solvent studies of the stability of DNA,RNA,and phosphoramidate-DNA helices[J].Journal of the American Chemical Society,1998, 120:9401-9409.
[20]Hou T J,Zhu L L,Chen L R,et al.Studies on interactions between EGFR and 4-anilinoquinazoline inhibitors[J].Acta Chimica Sinica-Chinese Edition,2002,60:1023-1028.
[21]蒋勇军,曾敏,周先波,等.CDK2-抑制剂结合自由能计算[J].Acta Chimica Sinica,2004,62:1751-1754.
[22]Sharp K A,Honig B.Electrostatic interactions in macromolecules: theory and applications[J].Annual Review of Biophysics and Biophysical Chemistry,1990,19:301-332.
[23]Sitkoff D,Sharp K A,Honig B.Accurate calculation of hydration free energies using macroscopic solvent models[J].The Journal of Physical Chemistry,1994,98:1978-1988.
[24]Kottalam J,Case D.Langevin modes of macromolecules:applications to crambin and DNA hexamers[J].Biopolymers,1990,29: 1409-1421.
[25]Still W C,Tempczyk A,Hawley R C,et al.Semianalytical treatment of solvation for molecular mechanics and dynamics[J].Journal of the American Chemical Society,1990,112:6127-6129.
[26]Janssen B J C,Halff E F,Lambris J D,et al.Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition[J].Journal of Biological Chemistry,2007,282:29241-29247.
[27]Chong L T,Duan Y,Wang L,et al.Molecular dynamics and freeenergy calculations applied to affinity maturation in antibody 48G7 [J].Proceedings of the National Academy of Sciences,1999,96: 14330-14335.
[28]Novotny J,Bruccoleri R E,Davis M,et al.Empirical free energy calculations:a blind test and further improvements to the method 1 [J].Journal of Molecular Biology,1997,268:401-411.
[29]Sharp K A.Electrostatic interactions in hirudin-thrombin binding [J].Biophysical Chemistry,1996,61:37-49.
[30]马志,张峰,张慧晓,等.基于分子动力学的miRNA 3′端与Argonaute蛋白PAZ功能域相互作用的研究[J].大连海洋大学学报,2012,27(5):441-447.
[31]López de Victoria A,Gorham Jr R D,Bellows-Peterson M L,et al.A new generation of potent complement inhibitors of the compstatin family[J].Chemical Biology&Drug Design,2011,77:431-440.
Binding free energy calculation of complement protein C3c-inhibitor Compstatin interaction by MM/PBSA method
Abstract:The binding free energy of human C3c-Compstatin complex was calculated by an MM/PBSA method, and the key residues interactions were also investigated between MG4-MG5 binding domain of C3c and Compstatin by using energy decomposition method.The results showed that the calculated binding free energy(-8.06 kcal/mol)was in high agreement with the binding affinity(-6.72 kcal/mol)by the experimental essays,the total energy in the molecule(-177.24 kcal/mol)showing the maximal contribution to binding complement inhibitor,followed by vaccum electrostatic interaction(-108.74 kcal/mol)and Van der Waals interaction(-68.51 kcal/mol). The dynamic process of combination between C3c and the inhibitor together with the changes of the protein under the effect of combination was simulated as a complementary part in the crystal experiments.Our molecular dynamics stimulation proved the experiment with the structure information:Residue ARG459,ASP491,MET457 of C3c and TRP7,TRP4,HIS10 of Compstatin had major contribution to the binding free energy of the complex.The information above provides some insights into the based structural design of inhibitor of complement 3.
Key words:molecular dynamics;MM-PBSA/GBSA method;binding free energy;compliment C3c;Compstatin
中图分类号:R331
文献标志码:A
收稿日期:2012-06-18
基金项目:国家自然科学基金资助项目(3047132)
文章编号:2095-1388(2013)02-0154-06